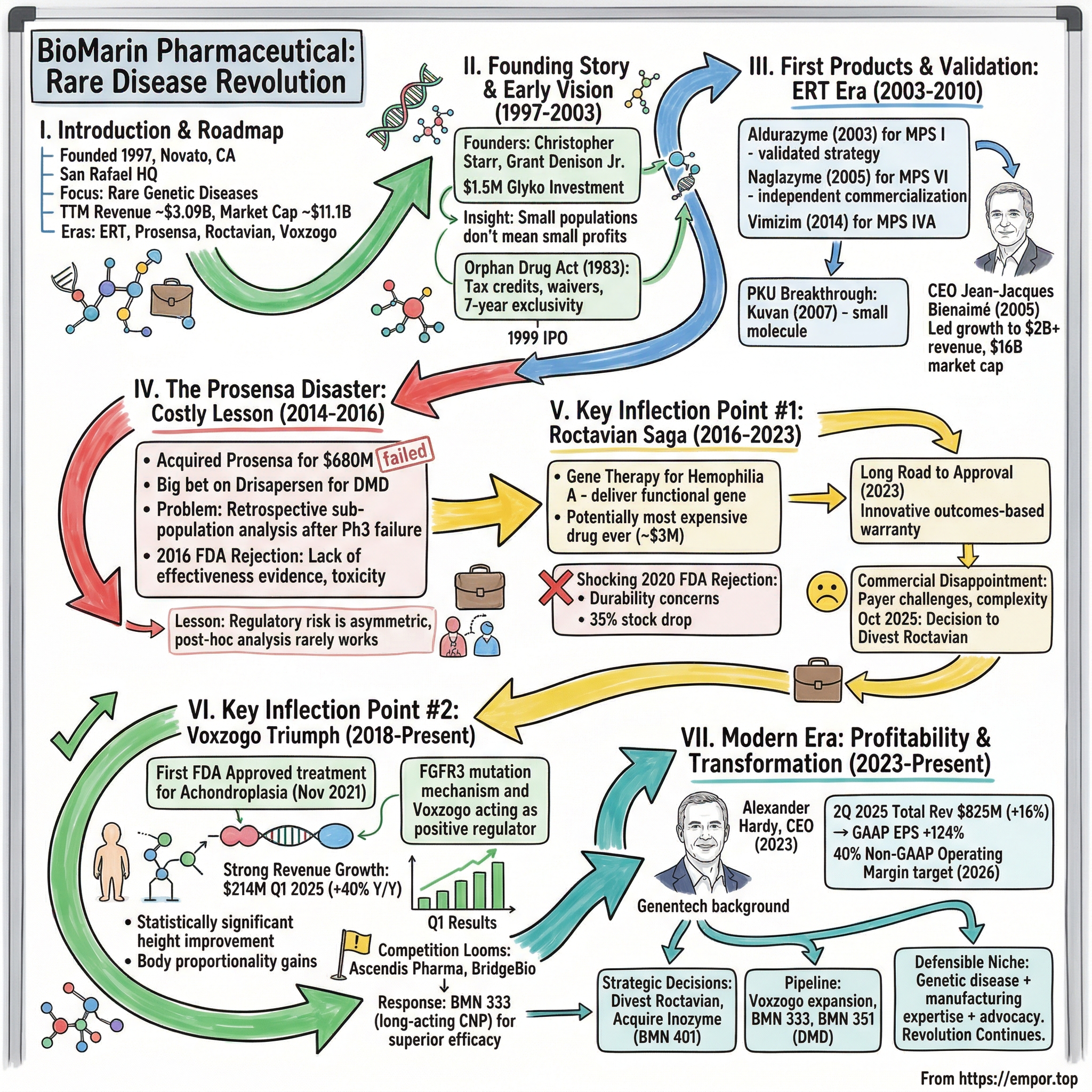
BioMarin Pharmaceutical: Pioneering the Rare Disease Revolution
I. Introduction & Episode Roadmap
The monsoon season arrives every year in San Rafael, California—but instead of tropical storms, the hills north of San Francisco witness a different kind of tempest: the relentless pursuit of cures for diseases that affect so few people that most pharmaceutical companies refuse to acknowledge their existence.
BioMarin Pharmaceutical Inc. is an American biotechnology company headquartered in San Rafael, California. With trailing twelve-month revenue of approximately $3.09 billion and a current market capitalization of roughly $11.1 billion, it stands as one of the rare success stories in orphan drug development—a company that bet everything on the forgotten patients and built an empire treating conditions most physicians will never encounter in their careers.
The core question that animates this story: How did a company founded in 1997 by Christopher Starr Ph.D. and Grant W. Denison Jr. with an investment of $1.5 million from Glyko Biomedical transform into the defining company in rare genetic diseases? The answer lies in a combination of regulatory arbitrage, scientific audacity, strategic missteps that nearly destroyed the company, and triumphant comebacks that validated two decades of patient work.
BioMarin Pharmaceutical Inc. is a global biotechnology company founded in 1997 in Novato, California, and headquartered in San Rafael, California, that discovers, develops, and commercializes therapies targeting rare and serious genetic diseases using expertise in genetics, enzyme technology, and molecular biology. The firm has brought eight marketed products to patients in over 80 countries.
This narrative unfolds across four distinct eras: the enzyme replacement therapy revolution of the early 2000s, the hubris of the Prosensa acquisition, the decade-long Roctavian saga that tested investor patience, and the current Voxzogo-driven resurgence that finally pushed the company to sustained profitability. Each chapter reveals something fundamental about the economics of orphan drugs, the politics of FDA approvals, and the delicate balance between scientific ambition and commercial reality.
What makes BioMarin's story particularly instructive is how it illuminates the paradox at the heart of rare disease drug development: "Orphan drugs are actually more profitable than non-orphan drugs... drug manufacturers are routinely able to price orphan drugs at US$100,000–US$200,000 per patient per year, needing only 5,000–10,000 patients to generate US$1 billion in annual revenues."
For investors seeking to understand the rare disease landscape, BioMarin represents both a cautionary tale and an inspiration—a company that has stumbled dramatically, recovered unexpectedly, and continues to pioneer treatments in therapeutic areas where it remains the unquestioned leader.
II. The Founding Story & Early Vision (1997–2003)
The Founders and the Insight
"When BioMarin first opened its doors back in 1997, treatment of rare disease was as rare as the conditions themselves."
The company's origin traces to the intersection of two careers. In 1997, Dr. Christopher Starr co-founded BioMarin Pharmaceutical Inc., serving as Senior Vice President and Chief Scientific Officer. At BioMarin, he led a scientific operations team to successfully develop commercial manufacturing processes for enzyme replacement products, and supervised the cGMP design, construction and licensing of a manufacturing facility. From 1991 to 1998, as Vice President of Research and Development at BioMarin Pharmaceutical Inc.'s predecessor company, Glyko, Inc., Dr. Starr supervised and directed research, development and commercial programs.
The founders, drawing on expertise in biotechnology and pharmaceuticals, aimed to pioneer treatments for rare genetic disorders, starting with enzyme replacement therapies for lysosomal storage diseases such as mucopolysaccharidosis. This focus addressed unmet needs in conditions affecting small patient populations, where traditional large-pharma incentives were limited due to low commercial potential.
This wasn't merely academic interest. The business model depended on a critical insight: small patient populations don't necessarily mean small profits if you can charge appropriately for life-changing treatments that have no alternatives. It's one thing to understand this intellectually; it's another to build a company around it when the regulatory framework barely exists and the market has yet to prove the economics work.
The Orphan Drug Act Tailwind
The foundation of BioMarin's business model rests on legislation signed fourteen years before the company's founding. On January 4, 1983, President Ronald Reagan signed the Orphan Drug Act into law. The Act is meant to facilitate development of orphan drugs—drugs for rare diseases such as Huntington's disease, myoclonus, ALS, Tourette syndrome or muscular dystrophy which affect small numbers of individuals residing in the United States.
The economic logic was elegant: The Kefauver-Harris Amendment dramatically increased the costs associated with developing new medicines. Pharmaceutical companies responded by focusing on developing treatments for common diseases in order to maximize the possibility of recouping research and development costs and generating significant profits. As a result, rare diseases were largely ignored due to poor economic potential and were thus said to be "orphaned."
The ODA changed the calculus fundamentally. The act offers tax credits, a waiver of the Prescription Drug User Fee, and extended market exclusivity options to participating drug developers. The Orphan Drug Approval Law of 1983 incentivized pharmaceutical companies to develop drugs for rare diseases by offering market exclusivity and tax credits.
The seven-year market exclusivity provision proved particularly important. For thirty-five years the Orphan Drug Act of 1983 has provided incentives for pharmaceutical manufacturers to develop drugs to treat rare diseases—conditions that affect fewer than 200,000 people in the US. One key statutory incentive is an exclusive seven-year marketing right for the rare disease indication, which has been heralded as driving a dramatic increase in the number of rare disease treatments.
The founders saw what others missed: the ODA created an asymmetric risk-reward profile. If you could develop a drug for a rare condition, you'd face minimal competition for years, command premium pricing, and build enduring relationships with patient communities desperate for any treatment. Before Congress enacted the ODA in 1983 only 38 drugs were approved in the USA specifically to treat orphan diseases. From the passage of the ODA in 1983 until May 2010, the FDA approved 353 orphan drugs and granted orphan designations to 2,116 compounds.
Early Strategic Moves
The acquisition of Glyko Biomedical in 1998 was significant for its focus on enzyme technology. BioMarin went public in 1999, securing capital for research and development.
The 1999 IPO occurred at a fortunate moment—the dot-com bubble was inflating, and investor appetite for speculative growth stories had never been higher. BioMarin completed its initial public offering. Since a large number of early investors were based in Europe, the company was listed on both the Swiss SWX Exchange and the Nasdaq National Market.
Seed investors were amongst others MPM Bioventures, Grosvenor Fund and Florian Schönharting. These weren't mainstream healthcare investors—they were specialists who understood the peculiar economics of orphan drugs and were willing to endure years of losses while the company built its scientific capabilities.
The early vision was narrow but deep: focus relentlessly on enzyme replacement therapies (ERTs) for lysosomal storage diseases. BioMarin's core business and research are in enzyme replacement therapies (ERTs). BioMarin was the first company to provide therapeutics for mucopolysaccharidosis type I (MPS I), by manufacturing laronidase (Aldurazyme, commercialized by Genzyme Corporation).
This specialization would prove prescient. Rather than competing with Big Pharma across multiple therapeutic areas, BioMarin carved out a niche so specialized that no major pharmaceutical company bothered to compete.
III. First Products & Validation: The ERT Era (2003–2010)
Aldurazyme: The First Win
Founded in Novato, California, in 1997, the company quickly expanded, acquiring Glyko Biomedical in 1998 and becoming a publicly traded company in 1999. The early 2000s saw the first FDA approval for Aldurazyme in 2003, followed by Naglazyme in 2005, marking the company's entry into the rare disease treatments market.
The Aldurazyme approval in 2003 was transformative—not because of the revenue it generated, but because it validated the scientific and regulatory strategy. BioMarin Pharmaceutical has pioneered enzyme replacement therapies (ERTs) for rare genetic disorders, particularly lysosomal storage diseases caused by enzyme deficiencies. The company's foundational work includes Aldurazyme (laronidase), approved by the FDA in 2003 for mucopolysaccharidosis type I (MPS I), which addresses alpha-L-iduronidase deficiency through intravenous infusion to reduce glycosaminoglycan accumulation.
The partnership with Genzyme for Aldurazyme commercialization taught BioMarin crucial lessons about market access and patient identification in rare diseases. Finding patients with MPS I—a condition affecting perhaps 1 in 100,000 births—required building relationships with academic medical centers, patient advocacy groups, and specialized physicians who might see one case in an entire career.
Building the ERT Portfolio
This was followed by Naglazyme (galsulfase) in 2005 for MPS VI, targeting N-acetylgalactosamine 4-sulfatase deficiency, and Vimizim (elosulfase alfa) in 2014 for MPS IVA (Morquio A syndrome), the first ERT specifically for N-acetylgalactosamine-6-sulfatase deficiency, demonstrating improved endurance in clinical trials via the 6-minute walk test. These therapies established BioMarin's expertise in recombinant enzyme production and delivery, with rapid progression from investigational new drug filing to approval in about five years for select programs.
Naglazyme's 2005 approval represented something different: BioMarin's first independently developed and commercialized medicine. The FDA approved BioMarin's first independently developed and commercialized medicine, and the first authorized treatment for a second form of mucopolysaccharidosis.
The economic model was proving itself. Each approved drug created a durable revenue stream with minimal competition. Patients diagnosed with these conditions had essentially no alternatives, and many would require treatment for the rest of their lives. The revenue per patient was extraordinary by pharmaceutical standards—often hundreds of thousands of dollars annually—but the small patient populations meant BioMarin would never become a true large-cap company through ERTs alone.
The PKU Breakthrough
BioMarin was also the first company to provide therapeutics for phenylketonuria (PKU).
Phenylketonuria represented a significant expansion of BioMarin's addressable market. Unlike the MPS conditions, PKU affects approximately 1 in 10,000 to 15,000 births—still rare, but an order of magnitude more common than MPS I or MPS VI. Kuvan's approval came in 2007, giving BioMarin its first product targeting a relatively larger rare disease population.
The strategic importance of PKU extended beyond immediate revenues. It demonstrated that BioMarin's capabilities could extend beyond enzyme replacement to other therapeutic modalities—in this case, a small molecule cofactor therapy.
Strategic Acquisitions
The late 2000s and early 2010s saw BioMarin begin building through acquisition. Further expansion came with strategic acquisitions like LEAD Therapeutics in 2010 and Zacharon Pharmaceuticals in 2012.
In 2012, BioMarin acquired Zacharon Pharmaceuticals, a private biotechnology company based in San Diego focused on developing small molecules targeting pathways of glycan metabolism. In 2014, BioMarin acquired a histone deacetylase inhibitor chemical library from Repligen for $2 million with the intention of advancing work toward therapies for Friedreich's ataxia and other neurological disorders.
These acquisitions were relatively modest in scale, but they signaled BioMarin's ambition to expand beyond its ERT foundation. The company was beginning to accumulate the scientific expertise and pipeline assets that would eventually diversify its revenue streams—though not always successfully, as the next chapter would demonstrate.
The arrival of Jean-Jacques Bienaimé as CEO in 2005 marked a turning point. Mr. Bienaimé joined BioMarin in May 2005, when the company had a single marketed product and approximately $26 million in annual revenues. Under Mr. Bienaimé's leadership, BioMarin has grown significantly and is expected to reach annual revenues of well over $2 billion in 2023.
He joined the company in May 2005 as Chief Executive Officer and member of the board of directors, bringing with him over 25 years of experience in the biotechnology and pharmaceutical industries. Under his leadership, the market capitalization of BioMarin went from around $450 million in May 2005 to approximately $16 billion in Summer 2016. BioMarin 2016 full-year revenues were $1.12 billion.
Bienaimé's background—including his tenure running Genencor—prepared him for the peculiar challenges of scaling a rare disease company. The playbook wasn't about mass marketing or primary care physician relationships; it was about building centers of excellence, supporting patient advocacy groups, and navigating the complex reimbursement landscape that orphan drugs inevitably face.
IV. The Prosensa Disaster: A Costly Lesson (2014–2016)
The Big Bet on Duchenne Muscular Dystrophy
In late 2014, BioMarin made the largest and most consequential bet in its history. BioMarin made a big bet on drisapersen in 2014 when it acquired Prosensa Holding N.V. for $680 million to gain access to its DMD assets, headlined by drisapersen.
The deal structure revealed management's confidence—and perhaps overconfidence. In November 2014, BioMarin Pharmaceutical agreed to pay up to $840 million to acquire Prosensa. In January 2016, the FDA rejected drisapersen, largely on the basis of toxicity.
BioMarin acquired Kyndrisa when it bought Prosensa of Leiden, Netherlands, in November 2014 for $680 million. In addition to the cash, there were two $80 million milestone payments related to regulatory approvals in the U.S. and Europe, which Prosensa shareholders will now not receive.
The strategic logic seemed compelling on the surface. Duchenne muscular dystrophy affects approximately 1 in 3,500 male births worldwide—far more common than BioMarin's existing patient populations. A successful DMD therapy could transform the company from a collection of small orphan drugs into a rare disease powerhouse with a potential blockbuster.
But there was a critical problem: Even though drisapersen had already failed a phase 3 study, prompting GlaxoSmithKline to back out of development, BioMarin thought it would be able to push forward with a retrospective analysis of sub-populations.
This was, in hindsight, extraordinary hubris. GlaxoSmithKline, with vastly more resources than BioMarin, had concluded the drug didn't work. BioMarin's thesis rested on the idea that post-hoc subgroup analysis would rescue a failed trial—a strategy that regulatory authorities view with deep skepticism.
The Unraveling
In November 2015, 15 of 17 members of the FDA's Peripheral and Central Nervous System Drugs Advisory Committee indicated they felt BioMarin's late-stage study lacked statistical significance. Of the three trials the company ran on Kyndrisa, two did not meet their statistical endpoint, which was getting patients taking the drug to walk farther in six months. This is a standard assessment for drugs being tested for similar diseases.
In January 2016, the FDA rejected drisapersen (Kyndrisa) after it concluded that the standard of substantial evidence of effectiveness had not been met.
The rejection wasn't merely a regulatory setback—it represented a fundamental repudiation of BioMarin's thesis for the acquisition. BioMarin had contended that the study was flawed for a variety of reasons, and that a post-hoc analysis pooling data from a number of subgroups showed that drisapersen really does benefit Duchenne patients. But that was an argument that panelists and FDA scientists didn't appear to buy.
Since then, the company has invested over $66 million into development for drisapersen, including $16.4 million in the first three months of 2016. Trouble began last fall, when an FDA advisory panel criticized the evidence backing up the drug. The FDA subsequently rejected the drug in January. BioMarin's last hope for drisapersen was in Europe, where it was still under regulatory review. But a May meeting with the key Committee for Medicinal Products for Human Use (CHMP) "clearly indicated" the panel would recommend rejection.
The Write-Off
Jean-Jacques Bienaimé, chairman and chief executive officer of BioMarin, said: "The withdrawal of the MAA and discontinuation of our current experimental drugs for Duchenne is a difficult but necessary decision at this time." The decision, while expected, is still another heavy blow to DMD patients and families.
In November 2014, the company agreed to the acquisition of Prosensa for up to $840 million; however, the range of treatments for Duchenne muscular dystrophy failed to attain FDA approval, and development ceased in May 2016.
Ending the program could result in a write-down of "all or a significant portion" of the costs related to BioMarin's acquisition of Prosensa. The company already recognized a nearly $200 million impairment charge to IPR&D assets tied to drisapersen in the fourth quarter of last year.
The Prosensa disaster offers several lessons for investors evaluating rare disease companies. First, regulatory risk in orphan drugs is asymmetric—when a trial fails, there's often nowhere to hide because you've typically bet the entire indication on a single pivotal study. Second, post-hoc subgroup analysis rarely survives regulatory scrutiny, regardless of how compelling the story appears. Third, acquiring a drug that has already failed Phase III represents a fundamentally different risk profile than acquiring a drug in earlier development—the latter preserves optionality while the former concentrates risk.
The silver lining, if there was one, came from the broader DMD competitive landscape. BioMarin's abandonment of Kyndrisa once again puts the focus on Sarepta Therapeutics. That company's DMD drug, eteplirsen, has been on a very dramatic roller-coaster ride. Sarepta ultimately secured approval, demonstrating that the FDA wasn't opposed to DMD therapies in principle—just to drugs without convincing evidence.
V. Key Inflection Point #1: The Roctavian Saga (2016–2023)
The Gene Therapy Moonshot
Even as the Prosensa disaster unfolded, BioMarin was quietly pursuing what it believed would be a transformative technology: gene therapy for hemophilia A. The concept was elegant—rather than requiring patients to receive regular infusions of clotting factor, why not deliver a functional copy of the gene itself and potentially cure the disease with a single treatment?
It was expected to be the most expensive drug ever approved by the Food and Drug Administration (FDA), at an estimated price tag of as much as $3 million.
The scientific premise was sound. Hemophilia A results from mutations in the gene encoding Factor VIII, a protein essential for blood clotting. Patients with severe hemophilia A face a lifetime of bleeding events and require regular prophylactic infusions costing, by BioMarin's calculation, approximately $25 million over a lifetime.
The regulatory path initially seemed favorable. The therapy, then known as valoctocogene roxaparvovec (later branded Roctavian), received breakthrough therapy designation from the FDA in 2017—a signal that the agency viewed it as a potentially important advance.
The Shocking 2020 FDA Rejection
Then came August 18, 2020—a date that BioMarin investors will not soon forget.
The Food and Drug Administration unexpectedly rejected two major medicines on Tuesday and Wednesday, turning back what would have been the first gene therapy for hemophilia as well as a closely followed anti-inflammatory treatment.
The FDA's decision not to approve a hemophilia gene therapy from BioMarin Pharmaceutical came as a shock to investors, who had expected the opposite based on months of company feedback. Shares of the Californian biotech fell 35% between Tuesday and Wednesday, wiping more than $7.5 billion from its market value.
In mid-afternoon trading, Biomarin Pharmaceutical Inc. shares plunged $42.62, or 36%, to $75.92. Trading volume by then was about 19 times the usual number of BioMarin shares traded in a day.
The rejection was particularly shocking because BioMarin's management had expressed confidence in approval. "We were quite confident," said company CEO Jean-Jacques Bienaime. "We had an agreement with the FDA over a year ago ... regarding the hurdle we'd have to pass for accelerated approval. Not only did they move the goalpost for accelerated approval, but they're now apparently moving the goalpost for full approval."
The Scientific Controversy
The FDA's concern centered on a fundamental question: how long would the gene therapy's effect last?
Three-year data on its candidate sparked concerns about the durability of the therapy after factor VIII levels seemed to fall off after 12 to 18 months, raising the possibility that patients might need to be re-dosed to maintain protection against bleeds.
In a letter to BioMarin, the FDA said it would need two-year follow-up data from a Phase 3 study of Roctavian. The California biotech estimates the last patient in that trial will complete two years of study in November 2021, likely setting up a resubmission and FDA review by mid- or late-2022. The requirement had not been specified by the agency previously, according to BioMarin.
But some in the medical community weren't surprised at all. "A lot of us were surprised, to be honest, that the FDA was allowing them to proceed in the first place," said Robert Sidonio, a pediatric hematologist at Children's Healthcare of Atlanta.
The discrepancy between Phase 1/2 and Phase 3 results created fundamental uncertainty. BioMarin had sought an accelerated approval for the therapy, relying on full results from a small study of about a dozen patients, coupled with an interim look at the first six months of the larger Phase 3 trial. The FDA ended up disagreeing, apparently concerned that patients in the Phase 3 study weren't faring as well as those in the earlier trial.
The Long Road to Approval
BioMarin spent the next two years gathering additional data and negotiating with the FDA. Before its initial rejection, the gene therapy scored a breakthrough therapy designation way back in 2017. But in 2020, BioMarin received a complete response letter from the FDA, with the agency voicing concerns over the duration of the drug's benefit. BioMarin resubmitted its latest application last September.
The drug, which snared a hemophilia green light as Roctavian in Europe this past August, is in the running to become the first gene therapy approved for the bleeding disorder in the U.S.
Finally, in June 2023, the FDA granted approval. Roctavian's approval in Europe in 2022 and in the U.S. a year later were scientific milestones, the culmination of years of research developing a genetic medicine for hemophilia A.
BioMarin priced the therapy at $2.9 million—not quite the $3 million that had been rumored, but still among the most expensive drugs ever approved. The company implemented an innovative outcomes-based warranty program to address payer concerns about durability.
Commercial Disappointment and Divestiture
The scientific triumph quickly became a commercial disappointment.
BioMarin quickly and sharply slashed its revenue forecasts for 2023 and ended up recording $3.5 million in product sales that year. The therapy accounted for only $26 million in 2024, and just $23 million over the first nine months of 2025.
In April, BioMarin predicted the therapy would rake in $50 to $150 million, but payer challenges have dogged its uptake. Eight months after gaining approval in Europe, Roctavian had yet to be taken by a single patient. And in the U.S., payer negotiations for the $2.9 million dollar therapy have also slowed its launch. Ultimately, Roctavian made under $1 million in the third quarter and BioMarin now estimates that it'll make less than $10 million this year.
CEO Alexander Hardy has previously cited the "complexity" of getting patients on treatment as a reason for Roctavian's commercial performance. But doubts about the durability of its benefits and a price tag that made reimbursement discussions challenging also slowed its sales trajectory.
In October 2025, BioMarin announced the inevitable conclusion: BioMarin Pharmaceutical is giving up on the hemophilia gene therapy Roctavian, announcing on Monday plans to offload a first-of-its-kind medicine once expected to become a future blockbuster. CEO Alexander Hardy said BioMarin will "pursue options to divest Roctavian and remove it from our portfolio."
Roctavian has become a cautionary tale of the challenges drugmakers can face selling a gene therapy. BioMarin wasn't alone in reporting sluggish sales, either. Pfizer cited weak demand in choosing to stop selling a gene therapy for the less common hemophilia B. Uptake has also been slow for CSL's Hemgenix, another hemophilia B gene therapy.
The Roctavian story illustrates a broader lesson about gene therapy commercialization: scientific success does not guarantee commercial success. The challenges of pricing a one-time cure, convincing payers to front-load payments, and overcoming patient hesitation about novel technologies can doom even scientifically validated therapies.
VI. Key Inflection Point #2: The Voxzogo Triumph (2018–Present)
A New Hope: Achondroplasia Treatment
While Roctavian captured headlines and investor attention, a quieter program was building toward what would become BioMarin's most important commercial success.
Voxzogo is the first FDA approved treatment for children with achondroplasia. BioMarin Pharmaceutical Inc. today announced that the U.S. Food and Drug Administration (FDA) has granted accelerated approval to VOXZOGO (vosoritide) for Injection, indicated to increase linear growth in pediatric patients with achondroplasia five years of age and older with open epiphyses (growth plates).
The approval came in November 2021, just as the Roctavian saga was entering its darkest chapter. FDA Approved: Yes (First approved November 19, 2021).
The worldwide incidence rate of achondroplasia is about one in 25,000 live births. While this qualifies as a rare disease under FDA definitions, it represents a significantly larger patient population than many of BioMarin's other indications.
The Science Behind the Success
In children with achondroplasia, endochondral bone growth, an essential process by which bone tissue is created, is negatively regulated due to a gain of function mutation in FGFR3. VOXZOGO, a C-type natriuretic peptide (CNP) analog, acts as a positive regulator of the signaling pathway downstream of FGFR3 to promote endochondral bone growth.
Unlike enzyme replacement therapies, which address the absence of a specific enzyme, Voxzogo works by counteracting an overactive signaling pathway. The drug doesn't cure achondroplasia, but it promotes more normal bone growth during the critical developmental years when children's growth plates remain open.
Baseline mean AGV in the placebo and Voxzogo groups was 4.06 cm/year and 4.26 cm/year, respectively. At week 52, the change from baseline in AGV was -0.17 cm/year for the placebo treated patients and 1.40 cm/year for the Voxzogo treated patients, resulting in a statistically significant improvement in AGV of 1.57 cm/year in favor of Voxzogo.
Expanding the Franchise
Previously, VOXZOGO was indicated for children who were 5 years of age and older. This expanded indication now includes children of all ages with open growth plates.
As of the end of the quarter, children with achondroplasia in 51 countries around the world were being treated with VOXZOGO, representing strong progress towards the company's target of accessing more than 60 countries by 2027.
The commercial trajectory has exceeded expectations. In 2024, strong global demand drove 42% fourth quarter growth and 56% full-year growth for VOXZOGO revenues, compared to the same periods in 2023.
BioMarin reported total revenues of $745 million for Q1 2025, representing a 15% increase compared to the same period last year. This growth was primarily driven by VOXZOGO, the company's treatment for achondroplasia, which generated $214 million in revenue, up 40% year-over-year.
Clinical Results That Matter
The real-world data continues to support the drug's value proposition. Growth improvements extend beyond simple height increases to include body proportionality and quality of life measures.
Children with achondroplasia treated with VOXZOGO experienced meaningful improvements in addition to height, such as gains in health-related quality of life (HRQoL), and increased bone length while maintaining bone strength.
With three years of follow-up, VOXZOGO-treated children had statistically significant improvements in body proportionality compared with untreated children of the same age range and gender. VOXZOGO is the only treatment for achondroplasia to have demonstrated, and been published in a peer-reviewed journal, statistically significant improvement in proportionality versus an untreated control arm.
But competition looms. Now, nearly four years later, the achondroplasia community is once again awaiting regulatory action that could open up a new treatment option for the disease. On November 30, the FDA is set to make a decision on Ascendis Pharma's TransCon CNP, which the biotech is proposing as a weekly treatment for children with achondroplasia.
"When Ascendis Pharma announced TransCon CNP results, BioMarin's stock fell 17%," highlighting investor concern about competition to Voxzogo's market position.
BioMarin is responding with its own next-generation therapy: BMN 333, our long-acting CNP, achieved our targeted profile in the healthy volunteer study and is now expected to move into the pivotal study in 2026. Our goal is for BMN 333 to demonstrate superiority to VOXZOGO and set a new standard for the treatment of achondroplasia.
VII. Modern Era: Profitability Inflection & Portfolio Transformation (2023–Present)
The Leadership Transition
Alexander Hardy has been named president and chief executive officer, succeeding Jean-Jacques Bienaimé, who will retire as chairman and CEO effective December 1, 2023. Mr. Hardy, currently the CEO of Genentech, will also serve as a director on BioMarin's board of directors.
Mr. Hardy, 55, brings more than 30 years of experience in the global healthcare and biotechnology industries, most recently serving as CEO of Genentech.
During his tenure, Bienaimé has steered BioMarin from a company with a single product and annual revenues of $26 million to a company with eight brands on the market, and revenues predicted to approach $3 billion in 2024.
Hardy's background at Genentech—where he oversaw the launch of 10 innovative medicines—brought exactly the commercial execution capability that BioMarin needed as Voxzogo scaled and Roctavian struggled.
The $4 billion goal would double 2023's total revenue of $2.4 billion and put BioMarin in the top quartile of biopharma revenue growth, Hardy said. To get there, BioMarin will depend on three separate but connected pillars.
Financial Inflection Point
"We were very pleased with our second quarter performance across all aspects of the business, including strong growth, exciting pipeline progress, and delivery of our business development strategy," said Alexander Hardy, President and Chief Executive Officer of BioMarin. "In the quarter, global demand for BioMarin's innovative therapies resulted in double-digit year-over-year revenue growth and significant profitability expansion."
Second Quarter 2025 Total Revenues of $825 million (+16% Y/Y and +17% at Constant Currency Y/Y), Second Quarter 2025 GAAP Diluted Earnings Per Share (EPS) of $1.23 (+124% Y/Y), Second Quarter 2025 Non-GAAP Diluted EPS of $1.44 (+50% Y/Y).
BioMarin's profitability metrics showed substantial improvement in Q2 2025. GAAP operating margin expanded to 33.5%, an increase of 16.6 percentage points year-over-year, while non-GAAP operating margin reached 39.9%, up 8.7 percentage points. GAAP diluted earnings per share more than doubled to $1.23, representing a 124% increase from $0.55 in Q2 2024. Non-GAAP diluted EPS grew 50% to $1.44. The company noted that EPS increased at more than three times the rate of revenue growth, reflecting successful implementation of operational efficiencies.
In 2024, BioMarin delivered record performance across the business. Total revenues for the full-year 2024 grew 18%. BioMarin's full-year GAAP Operating Margin of 17.0% expanded 9.3 percentage points Y/Y while GAAP Diluted EPS of $2.21 increased 154% Y/Y.
Strategic Portfolio Decisions
As a result of the completed acquisition of Inozyme on July 1, 2025, BioMarin expects to account for the transaction as an asset purchase and record the impact of acquired in-process research and development (IPR&D) charges in its third quarter 2025 financial results.
In July 2025, BioMarin completed the acquisition of Inozyme, adding BMN 401 (formerly INZ-701), a potential first-in-disease treatment for ENPP1 Deficiency, to BioMarin's Enzyme Therapies portfolio. Initial pivotal data readout for the ENERGY 3 study in children ages 1–12 years is anticipated in the first half of 2026, with potential launch in 2027. BioMarin is committed to continue working with the patient and HCP communities to identify individuals with ENPP1 Deficiency who may benefit from treatment with BMN 401.
The Roctavian divestiture decision reflects Hardy's willingness to make difficult portfolio choices. "As we focus on the business units aligned with our strategic priorities, today we are announcing the decision to pursue options to divest ROCTAVIAN and remove it from our portfolio. Today, the company announced its plan to pursue options to divest ROCTAVIAN, including exploring out-licensing opportunities."
Pipeline and Future Growth Drivers
BioMarin announced today that Phase 1 data in its healthy volunteer study with BMN 333, BioMarin's long-acting C-type natriuretic peptide (CNP), demonstrated area-under-the-curve (AUC) pharmacokinetic (PK) levels greater than three times the levels observed in other long-acting CNP studies. No safety signals were noted. Based on these results, BioMarin is advancing plans to initiate the registration-enabling Phase 2/3 study in the first half of 2026. BMN 333 is on track for a potential 2030 launch, should data be supportive.
In April 2025, BioMarin completed enrollment in its pivotal Phase 3 study with VOXZOGO in hypochondroplasia and the company is on track to share topline data in 2026, with a potential launch in 2027. BioMarin plans to leverage its multiyear track record treating children with achondroplasia, a related condition, to raise awareness and treat children with hypochondroplasia across the globe. The CANOPY clinical program is continuing to advance VOXZOGO in additional new indications, including idiopathic short stature, Noonan syndrome, Turner syndrome, and SHOX deficiency.
In 2025, the company will continue to implement additional components of the $500 million cost transformation program announced in September of 2024, with full realization of benefits expected in 2026. This ongoing integration of efficiencies in 2025 is expected to enable BioMarin to realize 40% Non-GAAP Operating Margin in 2026.
VIII. Controversies & Ethical Debates
Drug Pricing Criticism
Over the years, BioMarin has been criticised for drug pricing and for specific instances of denying access to drugs in clinical trials.
In April 2019, the BBC reported that patients who took part in a trial treatment for the drug Kuvan (sapropterin hydrochloride) were later denied access to it. The company was criticised by the NHS and Stephen Hammond MP for patient profiteering.
The company commented in response: "BioMarin is disappointed that the NHS England has not recognised the value of treating PKU patients with Kuvan, despite more than a decade of positive patient outcomes across 26 countries in Europe, Russia and Turkey."
Expanded Access Controversies
In 2013, BioMarin Pharmaceuticals was at the center of a high profile debate regarding expanded access of cancer patients to experimental drugs. On the advice of her doctor, Andrea Sloan, a patient with advanced ovarian cancer, requested that the company provide her with access to BMN 673, an unapproved PARP inhibitor drug candidate that had exhibited promising activity in a small Phase 1 clinical trial. The company declined, citing safety concerns.
In June 2019, a Belgian court ordered BioMarin to continue supplying Vimizim to a young girl suffering from Morquio syndrome free of charge. BioMarin stopped providing free Vimizim at the beginning of the year after negotiations with Belgian health authorities regarding reimbursement of the product repeatedly failed.
The Orphan Drug Paradox
The fundamental tension in orphan drug economics creates ongoing ethical challenges. "Orphan drugs are actually more profitable than non-orphan drugs." Critics argue that the ODA's incentives have been exploited to generate excessive profits from vulnerable patient populations.
Critics have questioned whether orphan drug legislation was the real cause of this increase, claiming that many of the new drugs were for disorders which were already being researched anyway, and would have had drugs developed regardless of the legislation, and whether the ODA has truly stimulated the production of non-profitable drugs; the act also has been criticised for allowing some pharmaceutical companies to make a large profit off drugs which have a small market but sell for a high price.
BioMarin's response to these criticisms emphasizes the lifetime value delivered to patients. A treatment that allows a child with achondroplasia to achieve more normal height and body proportions, or that prevents bleeding events in a hemophilia patient, represents a permanent improvement in quality of life. The question of whether the price accurately reflects this value—versus simply extracting maximum willingness-to-pay from healthcare systems—remains contested.
IX. Business & Competitive Analysis: Porter's 5 Forces & Hamilton's 7 Powers
Porter's Five Forces Analysis
1. Threat of New Entrants: LOW-MEDIUM
The barriers to entering BioMarin's markets are substantial but not insurmountable. Orphan drug designations provide seven years of market exclusivity, creating a significant first-mover advantage. The specialized knowledge required for enzyme replacement therapy manufacturing, patient identification, and rare disease clinical trials raises entry barriers further.
However, the Ascendis Pharma challenge to Voxzogo demonstrates that barriers aren't absolute. Well-funded competitors with differentiated approaches can target established franchises.
2. Bargaining Power of Suppliers: LOW
BioMarin's manufacturing expertise represents a core competency. The company has invested heavily in specialized facilities, reducing dependence on external manufacturing partners for its most important products.
3. Bargaining Power of Buyers: MEDIUM
Healthcare payers increasingly push back on orphan drug pricing, but patients with severe genetic diseases have essentially no alternatives. This creates tension: payers have significant negotiating leverage in aggregate, but for any individual patient, the drug is often either available or not available.
The Roctavian experience demonstrated that even approved drugs can fail commercially if payer negotiations prove intractable.
4. Threat of Substitutes: LOW (currently), INCREASING
For most of BioMarin's products, there are no direct substitutes. A patient with MPS VI has no alternative to Naglazyme. However, gene therapies theoretically represent a substitute threat to enzyme replacement therapies—if a one-time gene therapy could cure a condition, the lifetime value of an ERT franchise would evaporate.
5. Competitive Rivalry: LOW to MEDIUM
Within established indications, BioMarin faces minimal competition. The company's first-mover advantage in multiple rare diseases has created durable franchises. However, competition is intensifying in achondroplasia (Ascendis, BridgeBio) and could emerge in other indications.
Hamilton's 7 Powers Framework
1. Scale Economies: LIMITED
BioMarin's rare disease focus inherently limits scale advantages. Manufacturing volumes remain relatively small, and the company cannot achieve the cost advantages available to companies producing blockbuster drugs for large populations.
2. Network Effects: MINIMAL
Rare disease treatments don't benefit from traditional network effects. However, BioMarin's relationships with patient advocacy groups and centers of excellence create a form of information network that aids patient identification and market development.
3. Counter-Positioning: STRONG
BioMarin's exclusive focus on rare genetic diseases represents classic counter-positioning. Large pharmaceutical companies face internal conflicts when considering rare disease investments—the returns don't move the needle for their massive revenue bases, and the specialized expertise required doesn't leverage their existing capabilities.
4. Switching Costs: HIGH
For patients on enzyme replacement therapies, switching costs are extremely high. These are life-sustaining treatments, and patients who have stabilized on a particular therapy have strong reasons to continue.
5. Brand: MODERATE
BioMarin has built strong brand recognition within the rare disease community. Physicians, patient advocacy groups, and families trust the company based on decades of commitment to their conditions.
6. Cornered Resource: MODERATE
The company's accumulated expertise in rare disease drug development, manufacturing, and commercialization represents a cornered resource. This knowledge base is difficult to replicate quickly.
7. Process Power: MODERATE
BioMarin has developed specialized processes for rare disease drug development that enable faster progression from discovery to approval than would be typical for companies new to the space.
Competitive Positioning
BioMarin occupies a distinctive position in the biopharmaceutical landscape. It's too small and specialized to be considered a true large-cap pharmaceutical company, but too established and profitable to be classified with developmental biotech companies.
The competitive threat varies dramatically by product:
- Enzyme Replacement Therapies: Minimal competition; durable franchises with high switching costs
- Voxzogo: Intensifying competition from Ascendis Pharma; market leadership position currently strong but requires ongoing innovation (BMN 333) to maintain
- Pipeline: Mixed; success dependent on clinical outcomes in multiple programs
X. Bull Case vs. Bear Case
The Bull Case
1. Voxzogo's Expansion Potential
The CANOPY clinical program is testing Voxzogo in multiple additional indications beyond achondroplasia, including hypochondroplasia, idiopathic short stature, Noonan syndrome, Turner syndrome, and SHOX deficiency. BioMarin plans to leverage its multiyear track record treating children with achondroplasia, a related condition, to raise awareness and treat children with hypochondroplasia across the globe. The CANOPY clinical program is continuing to advance VOXZOGO in additional new indications.
If multiple indications receive approval, Voxzogo's addressable market could expand severalfold, potentially justifying a much higher valuation.
2. BMN 333 as a Competitive Moat Builder
Chief R&D Officer Greg Friberg underscored the potential of BMN-333, stating, "Our goal is clear: BMN-333 is designed to deliver superior efficacy versus Voxzogo."
By developing a next-generation therapy that could be superior to its own market-leading product, BioMarin is attempting to self-cannibalize before competitors can. If successful, this strategy could extend the company's leadership in skeletal conditions for another decade.
3. Profitability Inflection
This ongoing integration of efficiencies in 2025 is expected to enable BioMarin to realize 40% Non-GAAP Operating Margin in 2026.
The company's margin expansion has been dramatic, and the path to 40% operating margins would make BioMarin among the most profitable specialty pharmaceutical companies.
4. Pipeline Optionality
This year, we expect a number of innovative pipeline candidates to advance, including BMN 351 for Duchenne Muscular Dystrophy, and BMN 333 for multiple skeletal conditions, that will yield early clinical data read-outs.
Multiple pipeline programs create optionality that current valuation may not fully reflect.
The Bear Case
1. Voxzogo Competition
"The impact of potential Voxzogo competition heavily influenced BioMarin's decision to withdraw its revenue target," chief financial officer Brian Mueller said on the call.
Voxzogo currently reigns as the only therapy for achondroplasia, the most common cause of dwarfism, but two competitors are close on its heels: Ascendis Pharma, which has a target action date of Nov. 30 for TransCon CNP, and BridgeBio, which is developing a small-molecule drug that addresses the root molecular cause of the condition. BioMarin has now adjusted its 2027 top-line outlook to account for these competitors, admitting that the reign of Voxzogo may be nearing its end.
The entry of Ascendis and potentially BridgeBio could erode Voxzogo's market share and pricing power.
2. Gene Therapy Failure
The Roctavian experience demonstrates that even scientifically successful gene therapies can fail commercially. This outcome raises questions about BioMarin's strategic positioning in gene therapy more broadly.
3. Pipeline Concentration Risk
Much of BioMarin's growth story depends on BMN 333 demonstrating superiority to Voxzogo—essentially betting that the company can obsolete its own successful product before competitors do. If BMN 333 fails to show superiority, BioMarin could find itself with an aging franchise and no next-generation replacement.
4. Orphan Drug Pricing Pressure
Political and regulatory pressure on pharmaceutical pricing continues to intensify. Orphan drugs have historically been somewhat insulated from these pressures, but this could change.
Myth vs. Reality
Myth: BioMarin's rare disease focus makes it a defensive investment. Reality: The company faces meaningful competitive threats to its largest product and depends on successful pipeline execution for continued growth.
Myth: Gene therapy represents BioMarin's future. Reality: The Roctavian experience has caused the company to retreat from gene therapy commercialization, at least for now.
Myth: Orphan drugs are immune to pricing pressure. Reality: The Belgian court case and NHS controversies demonstrate ongoing tension between pricing and access.
XI. Key Performance Indicators to Track
For investors monitoring BioMarin, three KPIs deserve particular attention:
1. Voxzogo Revenue Growth Rate and Geographic Expansion
VOXZOGO continues to be a key growth driver for BioMarin, with the company reaffirming its 2025 revenue outlook of $900-935 million. The drug is now available in 55 countries, with approximately 75% of year-to-date revenue generated outside the United States.
Track quarterly Voxzogo revenue, year-over-year growth rates, and new country launches. Deceleration in growth or loss of market share to Ascendis's TransCon CNP would signal competitive pressure.
2. Non-GAAP Operating Margin Progression
This ongoing integration of efficiencies in 2025 is expected to enable BioMarin to realize 40% Non-GAAP Operating Margin in 2026.
The company has committed to reaching 40% non-GAAP operating margin by 2026. Progress toward this target—or failure to achieve it—will signal management's execution capability.
3. Pipeline Milestones
Key upcoming catalysts include: - BMN 333 Phase 2/3 initiation (1H 2026) - Voxzogo hypochondroplasia topline data (1H 2026) - BMN 401 (ENPP1 Deficiency) pivotal data (1H 2026) - BMN 351 (DMD) proof-of-concept data (2H 2025)
Binary pipeline events create both risk and opportunity. Positive readouts could significantly expand the company's addressable market, while failures would remove optionality from the investment thesis.
XII. Conclusion: The Rare Disease Revolution Continues
Twenty-eight years after its founding, BioMarin stands at another inflection point. The company that pioneered enzyme replacement therapy for MPS conditions, stumbled badly with Prosensa, nearly transformed itself with Roctavian, and hit its stride with Voxzogo now faces the challenge of defending its market position while building the next generation of rare disease therapeutics.
"On behalf of the Board, I would like to acknowledge JJ's exceptional leadership over the past 18 years in growing BioMarin into a leading, diversified genetic disease company. He has overseen the development and commercialization of seven medicines and through this led the transformation of the company into a profitable, fully integrated global business."
The passing of the baton from Bienaimé to Hardy represents more than a leadership change—it signals a new strategic era focused on operational excellence and portfolio discipline rather than transformative acquisitions.
For long-term investors, BioMarin presents a complex risk-reward profile. The bull case depends on continued Voxzogo expansion, successful BMN 333 development, and achievement of operating margin targets. The bear case centers on competitive pressure, pipeline disappointments, and the inherent challenges of sustaining growth in small patient populations.
What remains constant is BioMarin's positioning in one of healthcare's most defensible niches: the intersection of rare genetic diseases, specialized manufacturing expertise, and patient advocacy relationships built over decades. The company that bet everything on the forgotten patients has created a franchise that, for all its setbacks, continues to deliver life-changing treatments to children and families who have no alternatives.
The rare disease revolution that BioMarin helped pioneer shows no signs of slowing. The question is whether BioMarin will continue to lead it.
 RSS Feed
RSS Feed Spotify
Spotify Apple Podcasts
Apple Podcasts Amazon Music
Amazon Music Audible
Audible YouTube
YouTube