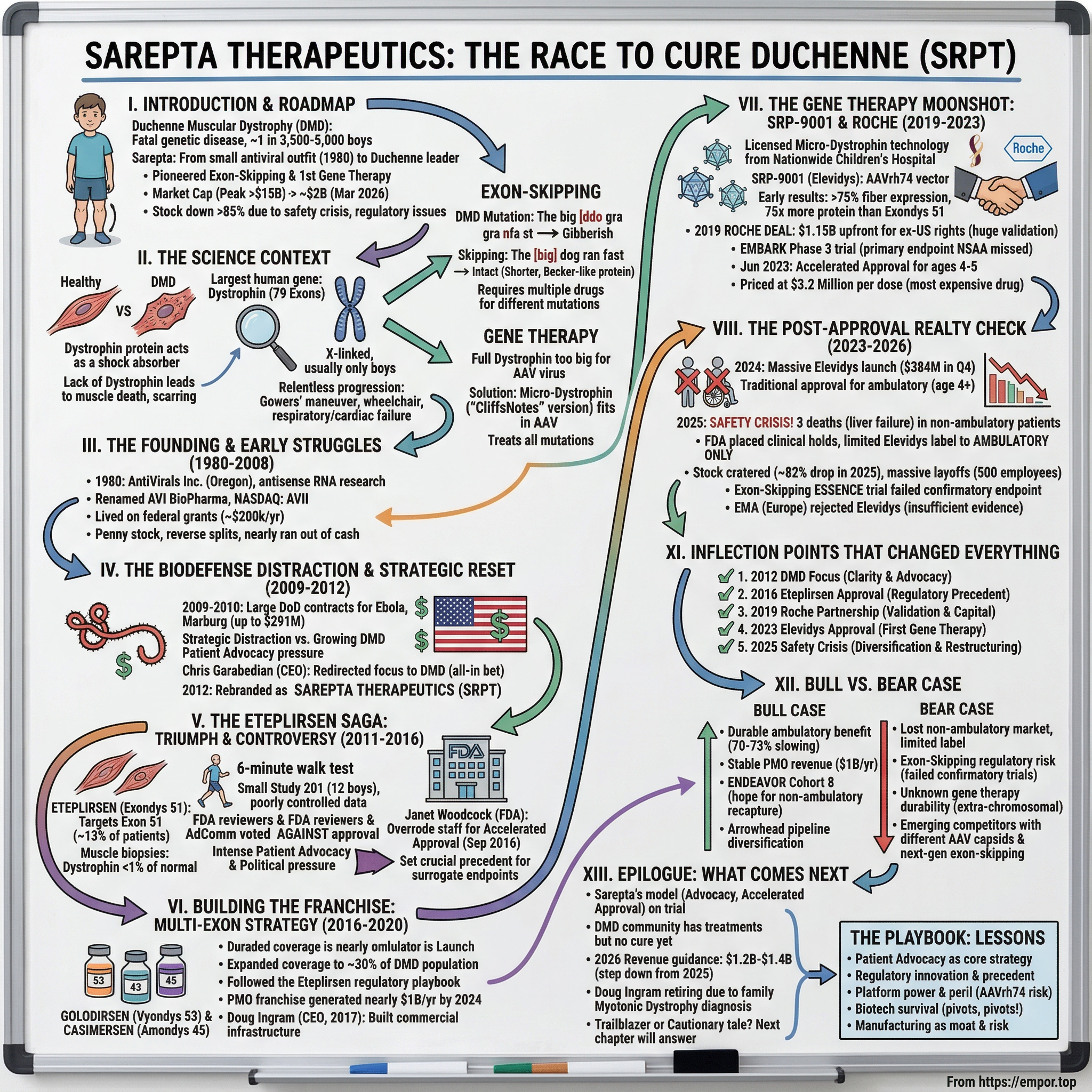
Sarepta Therapeutics: The Race to Cure Duchenne
I. Introduction and Episode Roadmap
Picture a six-year-old boy who trips and falls more than his classmates. His calves look oddly large, almost muscular, but the muscles are failing him. His parents notice he pushes himself up from the floor in a strange way, walking his hands up his own thighs just to stand. Within a few years, he will be in a wheelchair. Within two decades, without intervention, he will likely be dead. This is Duchenne muscular dystrophy, and for decades, the families who lived with it were told there was nothing medicine could do.
Sarepta Therapeutics is the company that refused to accept that answer. What began in 1980 as a small antiviral research outfit in Oregon has become the most important name in Duchenne treatment, pioneering both exon-skipping drugs and the first-ever gene therapy approved for the disease. At its peak, Sarepta commanded a market capitalization exceeding fifteen billion dollars. As of March 2026, that figure sits closer to two billion, battered by a safety crisis, regulatory upheaval, and the brutal realities of cutting-edge medicine. The company's stock has fallen more than eighty-five percent from its highs.
But the Sarepta story is not simply a biotech boom-and-bust narrative. It is a story about what happens when desperate families, controversial science, and the machinery of the FDA collide. It is a story about a company that nearly died half a dozen times before delivering a treatment that changed the trajectory of a fatal childhood disease. It is a story about the tension between statistical rigor and human urgency, between the scientific method and the moral imperative to act when children are dying.
Duchenne muscular dystrophy affects roughly one in every 3,500 to 5,000 boys born worldwide. It is caused by a single defective gene, making it theoretically addressable, but the gene in question is the largest in the entire human genome, which makes it extraordinarily difficult to fix. For most of medical history, the only treatment was corticosteroids, which slowed progression modestly but came with devastating side effects. Everything else was hope, fundraising, and waiting.
This episode covers the full arc. The founding and near-death experiences. The regulatory battles that rewrote FDA precedent. The scientific breakthroughs that produced partial and then potentially curative treatments. The massive Roche partnership that validated gene therapy. The 2023 approval that made history. The 2025 safety crisis that nearly destroyed the company. And the uncertain road ahead. Along the way, we will examine the business strategy, the competitive landscape, the manufacturing challenges, and the ethical questions that make Sarepta one of the most fascinating and polarizing companies in all of biotech.
The themes are timeless: patient advocacy as a force in drug regulation, the biotech survival playbook of pivots and near-death financing, platform science versus single-product bets, and the agonizing question of how society values treatments for rare, devastating diseases. Whether Sarepta ultimately succeeds or fails, the playbook it wrote will influence rare disease drug development for a generation.
II. The Science Context: What Is Duchenne and Why It Matters
To understand Sarepta, you first have to understand the disease it set out to conquer. Duchenne muscular dystrophy is not just any rare disease. It is the most common fatal genetic disorder of childhood, and its cruelty lies in its relentless progression.
DMD is caused by mutations in the gene that encodes a protein called dystrophin. Think of dystrophin as the internal scaffolding of muscle cells. Every time a muscle contracts, it generates enormous mechanical force, and dystrophin acts like a shock absorber, linking the internal skeleton of the cell to its outer membrane. Without dystrophin, the membrane tears with every contraction, and the muscle cell slowly dies. The body tries to repair the damage, but eventually the repair mechanism is overwhelmed, and muscle tissue is replaced by fat and scar tissue.
The dystrophin gene sits on the X chromosome, which is why DMD almost exclusively affects boys. Girls carry two X chromosomes, so even if one copy of the gene is defective, the other typically compensates. Boys have only one X chromosome, so a single bad copy means no functional dystrophin at all. About thirty percent of cases are spontaneous mutations with no family history, which means there is often no warning before the diagnosis.
The gene itself is a record-breaker. Spanning roughly 2.2 million base pairs and containing 79 exons, the dystrophin gene is the largest known gene in the human genome. It is so massive that it takes a cell approximately sixteen hours just to transcribe it from start to finish. This size is both the source of the problem and, paradoxically, part of why it became addressable. With 79 exons, there are many places where things can go wrong, but there are also many places where clever molecular tricks can work around the damage.
The typical progression is devastating and predictable. Symptoms usually appear between ages two and three: frequent falls, difficulty running, trouble climbing stairs, the characteristic Gowers' maneuver of walking hands up the thighs to stand. The calf muscles appear enlarged, a cruel illusion caused by fatty infiltration rather than strength. Without treatment, most boys lose the ability to walk independently by age nine or ten.
Corticosteroids, particularly deflazacort and prednisone, can extend ambulation by two to three years, but the decline continues and the side effects are severe: weight gain, bone fragility, mood disturbances, growth suppression. Scoliosis worsens rapidly once a child is wheelchair-bound, often requiring spinal fusion surgery. Respiratory muscles weaken, requiring ventilatory support by the mid-teens, first at night and eventually full-time. Cardiac muscle deteriorates, and dilated cardiomyopathy becomes nearly universal by the late teens.
Historically, life expectancy was the mid-twenties. Modern respiratory and cardiac care has extended that into the thirties and occasionally the forties, but the disease remains fatal. There is no spontaneous recovery, no remission, no plateau where the decline simply stops. Every year is worse than the last, and every lost capability is gone permanently.
What made DMD a testing ground for precision medicine was not just the severity of the disease but the elegance of the underlying biology. The body reads genetic code in groups of three nucleotides, called codons. When a mutation deletes one or more exons in a way that disrupts this three-letter reading frame, every downstream instruction becomes garbled, and no dystrophin is produced at all. This is Duchenne. But there is a milder cousin called Becker muscular dystrophy, where the deletion happens to preserve the reading frame, producing a shorter but partially functional protein. Becker patients often walk into their forties or fifties.
This observation was the scientific foundation for two revolutionary treatment strategies. The first was exon-skipping: using synthetic molecules to trick the cell's machinery into skipping over the damaged section of the gene, restoring the reading frame and producing a Becker-like shortened dystrophin. The second was gene therapy: delivering a miniaturized but functional version of the dystrophin gene directly into muscle cells using a harmless virus as a delivery vehicle.
Both approaches faced enormous technical challenges. Exon-skipping required drugs that could be delivered systemically, reach muscles throughout the body, and produce enough dystrophin to make a clinical difference. Gene therapy required fitting a functional gene into a viral vector with strict size limits, achieving body-wide distribution, and avoiding immune reactions. But the scientific community recognized that DMD, with its single-gene cause, known biology, and measurable biomarkers, was as close to an ideal target for genetic medicine as existed in nature.
The patient community recognized something else: that their children were dying, that the science was promising, and that the traditional pace of drug development was not fast enough. This collision of desperate parents, emerging science, and regulatory caution would define the next two decades of Sarepta's history.
To put the exon-skipping concept in the simplest possible terms, imagine the dystrophin gene as a sentence: "The big red dog ran fast." Now imagine a mutation deletes "red" but also shifts the spacing so the sentence reads "The big ddo gra nfa st." Complete gibberish, no functional meaning. That is Duchenne. Exon-skipping works by also deleting "big," so now the sentence reads "The dog ran fast." It is shorter than the original, it is missing some description, but the core meaning is intact. That is Becker. The genius of the approach is that you are not adding anything new; you are selectively removing just enough to restore the reading frame and let the cell produce a shortened but functional protein. The limitation is that each mutation requires a different "word" to be removed, which is why different drugs are needed for different exons.
The gene therapy approach takes a completely different tack. Rather than trying to fix the patient's broken instruction manual, you deliver an entirely new manual, albeit an abridged edition. The challenge is that the delivery vehicle, a harmless virus called AAV, can only carry a manual about half the size of the original. So scientists created a "CliffsNotes" version of dystrophin, keeping only the most essential sections. This micro-dystrophin is smaller but retains the structural function that muscle cells need to survive. And because the replacement gene works regardless of what specific mutation broke the original gene, a single gene therapy product can theoretically treat all DMD patients, not just the thirteen percent or eight percent eligible for a particular exon-skipping drug.
Understanding these two strategies, their respective strengths and limitations, is essential context for everything that follows in the Sarepta story. The company would pursue both approaches simultaneously, with exon-skipping providing the first beachhead and gene therapy representing the ultimate ambition.
III. The Founding and Early Struggles (1980-2008)
The company that would become Sarepta Therapeutics was born in the least glamorous corner of the biotech universe. In 1980, a scientist named James Summerton incorporated AntiVirals Inc. in Corvallis, Oregon, a college town best known for Oregon State University's beavers, not for pharmaceutical innovation. Summerton's vision was to develop drugs that could interfere with viral replication using synthetic molecules that bound to RNA, a technology called antisense oligonucleotides that was then in its absolute infancy.
For most of its first two decades, AntiVirals survived on federal research grants averaging roughly two hundred thousand dollars a year. This was hand-to-mouth biotech, the kind of company that existed in academic papers and NIH grant applications rather than in pharmaceutical markets. The technology was genuinely novel, built around a unique chemical backbone called phosphorodiamidate morpholino oligomers, or PMOs, which Summerton's lab developed. PMOs were more stable and less toxic than earlier antisense chemistries, but they were solutions in search of a problem, and the company cycled through antiviral targets without producing a commercial product.
By the early 1990s, the company had renamed itself AVI BioPharma and was publicly traded on NASDAQ under the ticker AVII. The IPO in 1997 was less a victory lap than a survival mechanism, raising the capital needed to keep the lights on and the research going.
The biotech landscape of the late 1990s was littered with similar companies: promising science, no revenue, constant dilution, and the ever-present threat of running out of cash before running out of ideas. For every Amgen or Genentech that successfully transitioned from research outfit to commercial pharmaceutical company, dozens of small biotechs quietly ran out of money and disappeared.
The early 2000s brought a shift in focus but not in fortune. AVI BioPharma began exploring its PMO technology for Duchenne muscular dystrophy, forming academic partnerships that would eventually prove transformative. But the company was simultaneously pursuing antiviral applications, spreading its limited resources across too many programs. Quarterly net losses ran in the tens of millions, and the stock price reflected the market's skepticism. At various points, AVI BioPharma traded at penny-stock levels, requiring reverse stock splits to maintain its NASDAQ listing.
Throughout this period, the company occupied a peculiar position in the biotech ecosystem. Its underlying technology was genuinely interesting, particularly the PMO chemistry that offered advantages over competing antisense approaches. Academic researchers studying DMD had begun to recognize that exon-skipping could theoretically convert severe Duchenne mutations into milder Becker-like forms. Groups at academic centers in the Netherlands, the United Kingdom, and Japan were publishing proof-of-concept studies showing that antisense oligonucleotides could indeed force exon skipping in cell cultures and animal models. The Dutch group, led by Judith van Deutekom and Annemieke Aartsma-Rus at Leiden University Medical Center, was particularly influential, publishing landmark papers that demonstrated the principle in human muscle cells. A competing company called Prosensa, later acquired by BioMarin, was developing a rival exon-skipping drug in the Netherlands.
But academic interest and commercial viability are very different things, and AVI BioPharma lacked the financial resources, the clinical infrastructure, and the strategic focus to bridge that gap. The company was caught in the biotech valley of death: too advanced for pure academic research, too early and too underfunded for commercial development. Its annual R&D spending was a fraction of what major pharmaceutical companies spent on a single clinical trial. The specialized neuromuscular disease centers that would need to administer and monitor exon-skipping treatments were scattered across the country, and building relationships with them required the kind of sustained commercial effort that a near-bankrupt company simply could not afford.
For investors who look at Sarepta's trajectory and see a straight line from vision to execution, the reality of this period is a useful corrective. The company spent nearly three decades wandering through scientific and commercial wilderness before finding its path. Most companies in similar positions simply cease to exist. That AVI BioPharma survived at all was partly luck, partly the stubbornness of scientists who believed in their technology, and partly the sheer difficulty of killing a company that has almost no fixed costs and just enough grant funding to keep a few researchers employed. The biotech graveyard is full of companies with better science and more resources that failed. AVI BioPharma's survival was its first miracle.
IV. The Biodefense Distraction and Strategic Reset (2009-2012)
By the late 2000s, AVI BioPharma was in existential crisis. The company had been publicly traded for over a decade without producing a single commercial product. Cash was burning at thirty to thirty-five million dollars per year. The DMD exon-skipping program was scientifically promising but years away from generating revenue. Something had to give.
The lifeline came from an unexpected source: the United States government. In 2009, AVI BioPharma secured an $11.5 million contract from the Defense Threat Reduction Agency. Then, in July 2010, the company landed a far larger prize: a contract worth up to $291 million from the Department of Defense for development of therapeutic candidates against Ebola and Marburg hemorrhagic fever viruses. This was not mere academic research funding. This was a massive government biodefense program that promised to keep the company solvent for years.
The biodefense work was a classic strategic distraction. It provided revenue and legitimacy, but it pulled management attention, scientific talent, and corporate resources away from the DMD program that represented the company's only realistic path to commercial value. Quarterly revenues from government contracts were modest, just a few million dollars, but the overhead of managing a complex government program in a small company was enormous.
Meanwhile, the DMD patient advocacy community was growing increasingly organized and impatient. Organizations like Parent Project Muscular Dystrophy and CureDuchenne, led by parents who had watched their sons deteriorate while waiting for treatments that never came, were raising awareness, funding research, and demanding action from both drug companies and regulators. These were not abstract stakeholders filing comment letters. These were mothers and fathers who could calculate, with terrible precision, how many months their sons had left before they would lose the ability to walk, and then how many years before they would lose the ability to breathe.
The pressure from patient advocates was both moral and practical. These families were raising millions of dollars for research, organizing congressional outreach, building registries of patients who would be willing to participate in clinical trials, and creating a political constituency for rare disease drug development that had never existed before. For a company like AVI BioPharma, which needed patients for trials and political support for regulatory pathways, ignoring these families was not an option.
The strategic reset came in stages. Chris Garabedian, who took over as CEO, began redirecting the company's focus toward DMD as the primary program. Garabedian was an outsider to the rare disease world, a former pharmaceutical executive who saw in AVI BioPharma's DMD program something that many investors had missed: a disease with a clearly defined genetic cause, a passionate and organized patient community, and an emerging regulatory framework for rare diseases that could compress development timelines. He was aggressive, sometimes abrasive, and completely convinced that DMD was the right bet.
The biodefense work continued but was deprioritized. The decision to go all-in on Duchenne was not obvious at the time. It meant betting the entire company on a single disease, a single technology platform, and a patient population of perhaps fifteen to twenty thousand people in the United States. If the science failed or the FDA said no, there would be no fallback. Garabedian's critics within the company worried that abandoning the biodefense revenue stream would leave Sarepta fatally exposed. His supporters argued that strategic focus was the only path to commercial value, and that trying to be two companies at once was a recipe for being neither.
On July 12, 2012, the transformation was made official. AVI BioPharma announced it had changed its name to Sarepta Therapeutics, with a simultaneous ticker change from AVII to SRPT. The rebrand was accompanied by a one-for-six reverse stock split, a standard maneuver for companies escaping penny-stock status. Garabedian described it as the beginning of an "important and exciting new phase," and for once, the CEO hyperbole turned out to be accurate.
The name itself, Sarepta, was drawn from a biblical reference to the ancient city where the prophet Elijah performed miracles. Whether or not the choice was intentional, the symbolism was apt: a company hoping to deliver miraculous treatments for a disease long considered untreatable.
The name change was more than cosmetic. It signaled to investors, to the FDA, and most importantly to the DMD community that this company was fully committed to their cause. The Ebola and Marburg programs would continue to wind down. The entire corporate strategy would revolve around using PMO technology to develop exon-skipping drugs for Duchenne. The company that had spent thirty-two years as a scientific wanderer had finally found its mission.
V. The Eteplirsen Saga: Triumph and Controversy (2011-2016)
The eteplirsen story is one of the most dramatic regulatory battles in FDA history, and understanding it is essential to understanding both Sarepta and the modern landscape of rare disease drug development.
Eteplirsen was Sarepta's first exon-skipping drug, designed to bind to exon 51 of the dystrophin pre-mRNA and cause it to be excluded during processing. In simple terms, imagine the dystrophin gene as a long instruction manual with seventy-nine chapters. In patients with certain mutations, a critical chapter is missing, and the resulting instructions are garbled beyond the point of the deletion. Exon-skipping works by covering up the chapter immediately adjacent to the gap, which forces the cell to skip it entirely and read straight through. The resulting manual is shorter but still makes sense, producing a truncated but partially functional protein.
Exon 51 is the most commonly targetable exon because many different deletion patterns can be rescued by skipping it, making roughly thirteen percent of DMD patients potentially eligible for treatment. It was the obvious place to start.
The clinical program that would determine eteplirsen's fate was remarkably small. Study 201 enrolled just twelve boys with Duchenne, aged seven to thirteen, randomized to receive eteplirsen at two different doses or placebo. For context, a typical Phase 3 drug trial enrolls hundreds or thousands of patients. Twelve is what most clinical trialists would call a "proof of concept" study, not a registration trial. But in DMD, where only about fifteen to twenty thousand patients existed in the entire United States and only a fraction had the right mutation for exon 51 skipping, large trials were impractical.
After twenty-four weeks, all participants enrolled in an open-label extension called Study 202, where everyone received the drug and was followed for up to four years. This meant that after the initial six months, there was no longer a true control group: every patient was being treated. Any comparison would have to be against external historical controls, a methodology that statisticians view with deep skepticism. The entire future of the company, and potentially the treatment paradigm for an entire disease, rested on what happened to these twelve boys.
The early results were intriguing but ambiguous, a pattern that would become a recurring theme in Sarepta's story. Muscle biopsies showed increased dystrophin-positive fibers, rising from roughly twenty-three percent at twenty-four weeks to over fifty percent at forty-eight weeks. On the six-minute walk test, a standard measure of functional ability, eteplirsen-treated boys appeared to maintain their walking ability better than historical controls, particularly when two patients with unusually rapid disease progression were excluded from the analysis. The numbers were encouraging, but the trial was too small and too poorly controlled to draw definitive conclusions.
Sarepta filed for FDA approval based on this data, and the regulatory drama began in earnest. The FDA's scientific reviewers were skeptical. Their analysis of the muscle biopsy data showed dystrophin levels of approximately 0.93 percent of normal, far below the threshold that most scientists believed was necessary for clinical benefit. One FDA reviewer, Dr. Ellis Unger, argued that dystrophin levels would need to reach at least ten percent of normal to confer meaningful benefit, more than ten times what eteplirsen appeared to produce. The trial design was criticized as fundamentally inadequate, with no proper control group for the functional endpoints.
In April 2016, the FDA convened its Peripheral and Central Nervous System Drugs Advisory Committee to review eteplirsen. What followed was one of the most emotionally charged advisory committee meetings in FDA history. The committee room was packed with DMD families. Parents testified about watching their sons lose the ability to walk, to feed themselves, to breathe independently. Advocacy organizations including CureDuchenne, led by Debra Miller, and Parent Project Muscular Dystrophy had mobilized thousands of families for letters, testimony, and lobbying.
The scientific vote was devastating. On whether there was substantial evidence that eteplirsen produced dystrophin at levels reasonably likely to predict clinical benefit, the committee voted six to seven against. On whether there was evidence of clinical effectiveness, the vote was three to seven against, with three abstentions. By traditional FDA standards, eteplirsen was dead.
But DMD is not a traditional disease, and 2016 was not a traditional regulatory moment. Congressional representatives from both parties had written letters to the FDA urging approval. In 2014 and 2015, multiple Congressional hearings had featured DMD families testifying about their children's deterioration while waiting for treatments. In one particularly powerful moment, a father described carrying his teenage son, who could no longer walk, up the stairs of their home every night, knowing that in a few years he would not even be able to sit up on his own. These testimonies were broadcast on C-SPAN and shared widely on social media, creating a level of public pressure that the FDA rarely experiences for a single drug application.
Patient advocacy groups had launched media campaigns, collecting hundreds of thousands of petition signatures and running advertisements in Washington, D.C. media markets. The Wall Street Journal, The New York Times, and STAT News all ran major features on the eteplirsen debate, framing it as a test case for how the FDA should balance scientific certainty against patient urgency. And within the FDA itself, a remarkable internal battle was unfolding.
Dr. Janet Woodcock, then director of the FDA's Center for Drug Evaluation and Research, believed that the evidence, while imperfect, was sufficient for accelerated approval. The accelerated approval pathway, designed for serious diseases with unmet medical need, allows drugs to be approved based on a surrogate endpoint that is "reasonably likely to predict" clinical benefit, with the requirement that confirmatory trials demonstrate actual benefit post-approval. Woodcock argued that the increase in dystrophin, however modest, met this standard in the context of a universally fatal disease with no alternative treatments.
Her own staff disagreed vehemently. Unger filed a formal internal appeal. Another senior FDA scientist, Dr. Luciana Borio, sided with Unger. The dispute was escalated to then-FDA Commissioner Robert Califf, who ultimately deferred to Woodcock's judgment.
It was an extraordinary situation: the head of drug evaluation overriding her own scientific staff and an external advisory committee to approve a drug that most of the FDA's internal experts believed had not met the evidentiary bar. Nothing quite like it had happened at the FDA in recent memory.
On September 19, 2016, the FDA granted accelerated approval to eteplirsen under the brand name Exondys 51. The approval letter contained a remarkable caveat: "A clinical benefit of Exondys 51, including improved motor function, has not been established." Sarepta was required to conduct confirmatory trials demonstrating actual clinical benefit, with the threat of approval withdrawal if those trials failed. The annual cost was set at approximately three hundred thousand dollars per patient.
The reaction was volcanic. DMD families celebrated, some weeping openly. Parent Project Muscular Dystrophy issued a triumphant statement. CureDuchenne's Debra Miller, whose own son Hawken had Duchenne, called it a turning point for the entire rare disease community. Scientists and FDA insiders agonized. Articles in JAMA and Nature Medicine called the approval a dangerous precedent. Dr. Aaron Kesselheim, a Harvard professor and FDA scholar, published a withering analysis arguing that the approval undermined the scientific foundations of drug regulation. Ellis Unger, the FDA reviewer who had opposed the decision, later told reporters that the study Sarepta had used in its filing was "misleading" and "should probably be retracted."
Supporters argued that in a disease where children lose function every month, the traditional pace of evidence gathering was itself harmful. The alternative to approving eteplirsen was not a better drug; it was nothing. Every month of delay meant more boys losing the ability to walk, and once lost, that function never returned. Critics countered that approving drugs without adequate evidence could undermine the entire regulatory framework and expose patients to ineffective treatments with real side effects. The annual cost of three hundred thousand dollars per patient made this more than an academic debate: insurance companies and government payers would be asked to spend hundreds of millions of dollars on a drug that the FDA's own approval letter admitted had not been shown to provide clinical benefit.
Sarepta's stock surged on the approval, vindicating investors who had held through the years of uncertainty. But the market's celebration obscured an uncomfortable truth: the drug had been approved with an explicit acknowledgment that clinical benefit was unproven, and the confirmatory trial obligation meant that the approval could theoretically be reversed if subsequent evidence was insufficient.
Whatever one's position on the science, the eteplirsen approval was a watershed moment. It demonstrated that organized patient advocacy could influence FDA decision-making at the highest levels. It established dystrophin production as an accepted surrogate endpoint for DMD drugs. And it created a regulatory template that Sarepta would use repeatedly over the next decade, for better and for worse. The drug approval also set a precedent that rippled across the rare disease landscape: if dystrophin production at less than one percent of normal could support approval in DMD, what biomarker thresholds would the FDA accept in other diseases?
VI. Building the Franchise: The Multi-Exon Strategy (2016-2020)
With Exondys 51 on the market, Sarepta moved quickly to expand its exon-skipping franchise. The strategic logic was compelling: eteplirsen addressed only the approximately thirteen percent of DMD patients with mutations amenable to exon 51 skipping. Different drugs targeting different exons could extend the platform to a much larger share of the patient population.
Golodirsen, targeting exon 53, was the second drug in the pipeline. It addressed roughly eight to ten percent of DMD patients. Sarepta submitted its application using the same regulatory playbook that had worked for eteplirsen: demonstrate dystrophin production via muscle biopsy as a surrogate endpoint, seek accelerated approval, and promise confirmatory trials later. But the path was not smooth. In August 2019, the FDA issued a Complete Response Letter, a formal rejection, citing concerns about infection risks related to intravenous infusion ports and kidney toxicity observed in animal studies at doses ten times higher than those used in humans.
Sarepta filed a formal dispute resolution request, and the concerns were resolved through a process led by Dr. Peter Stein, director of the FDA's Office of New Drugs. Just four months after the rejection, in December 2019, the FDA granted accelerated approval to golodirsen under the brand name Vyondys 53. The speed of the reversal surprised many observers and raised questions about whether the initial rejection had been justified or whether the FDA was simply moving through the motions of skepticism before granting what it had already decided to approve.
Casimersen, the third drug in the series, targeted exon 45 and covered approximately eight percent of DMD patients. It followed a more straightforward path, receiving accelerated approval in February 2021 under the brand name Amondys 45.
By this point, the regulatory playbook was well established: demonstrate dystrophin production via muscle biopsy, cite the precedent of the prior approvals, file under the accelerated approval pathway, and rely on the patient community and the urgency of the disease to overcome residual FDA skepticism. Each successive approval was incrementally easier than the last, as the precedent became more deeply established.
Together, the three exon-skipping drugs covered roughly thirty percent of the DMD patient population. Sarepta had built something genuinely unusual in biotech: a franchise of multiple drugs addressing different genetic subsets of a single disease, all using the same underlying technology platform. Each drug required its own clinical program and its own regulatory filing, but the scientific, manufacturing, and commercial infrastructure was shared.
Doug Ingram inherited a company with a single approved product generating modest revenue, a pipeline of two additional exon-skipping drugs in development, and an early-stage gene therapy program that was years from the clinic. His task was to build a real pharmaceutical company out of what had been, for most of its existence, a research organization with a commercial sideline. His Allergan experience proved invaluable: he understood how to build sales forces, navigate payer negotiations, manage regulatory relationships, and execute the operational blocking and tackling that separates promising biotech companies from successful pharmaceutical enterprises.
The commercial execution was harder than the science. DMD is a rare disease with a small, geographically dispersed patient population. Identifying eligible patients required genetic testing, specialist diagnosis, and coordination with neuromuscular disease centers. Reimbursement was complicated by the high price tags and the ongoing debate about whether the drugs actually worked. Insurance companies demanded prior authorization, imposed step therapy requirements, and in some cases simply denied coverage. Sarepta had to build an entire commercial infrastructure from scratch: a specialty sales force, a patient identification program, a reimbursement support team, and relationships with the handful of academic medical centers that treated most DMD patients.
Revenue grew steadily, from modest levels at Exondys 51's launch to a combined PMO franchise generating nearly a billion dollars annually by 2024. This was a remarkable commercial achievement for a set of drugs that the FDA's own approval letters acknowledged had not demonstrated clinical benefit. The revenue growth reflected both the desperation of the patient community and the effectiveness of Sarepta's commercial organization in navigating the labyrinthine world of rare disease reimbursement.
But the growth was accompanied by persistent questions about confirmatory evidence. The FDA had required post-approval trials to demonstrate actual clinical benefit for all three drugs, and those trials were proving difficult. The ESSENCE study, designed to confirm the clinical benefit of Vyondys 53 and Amondys 45, faced slow enrollment and challenging endpoint design. When ESSENCE ultimately reported results in late 2025, it failed to achieve statistical significance on its primary endpoint, leaving the confirmatory obligation unfulfilled.
This was the fundamental tension of Sarepta's exon-skipping franchise. The drugs were on the market, generating revenue, and being used by patients and families who believed they were beneficial. But the clinical evidence remained controversial, and the confirmatory trials that were supposed to resolve the debate kept falling short. Every drug that had previously failed its confirmatory trial after accelerated approval had been withdrawn from the market. Sarepta's exon-skipping drugs remained a live exception.
Meanwhile, Doug Ingram had arrived as CEO in June 2017, replacing Chris Garabedian. Ingram brought an entirely different profile to the role. A lawyer by training with a J.D. summa cum laude from the University of Arizona, he had spent nearly three decades at Allergan, rising to the position of President and helping navigate the company through its acquisition by Actavis. He was not a scientist, but colleagues described him as someone who understood the science deeply and communicated it with the conviction of a trial attorney. He was aggressive in his advocacy for Sarepta's drugs, intensely engaged with the FDA, and unapologetically public in his positioning as a champion of DMD patients. Under Ingram's leadership, Sarepta's revenue would grow at approximately forty percent compounded annually, and the company would evolve from a single-product exon-skipping company into a gene therapy pioneer.
VII. The Gene Therapy Moonshot: SRP-9001 and the Roche Partnership (2019-2023)
Even as the exon-skipping franchise expanded, Sarepta's leadership recognized its fundamental limitation. Exon-skipping produced only modest amounts of dystrophin. Each drug worked for only a specific subset of mutations. And the weekly intravenous infusions were a lifelong burden for patients and families. The real prize was gene therapy: a single treatment that could work for all DMD patients regardless of their specific mutation, delivering enough functional dystrophin to meaningfully slow or halt disease progression.
The scientific foundation came from Nationwide Children's Hospital in Columbus, Ohio, where two researchers had spent years developing a gene therapy approach for DMD. Dr. Jerry Mendell, a pioneering neurologist who had been working on neuromuscular disease for decades, and Dr. Louise Rodino-Klapac, a molecular biologist, had designed a clever solution to the fundamental packaging problem.
The challenge was this: the full-length dystrophin gene is approximately 11,200 base pairs long. The only proven delivery vehicle for systemic gene therapy, adeno-associated virus or AAV, can package approximately 4,700 base pairs. The gene is nearly two and a half times too large for the delivery system. You cannot simply shrink a virus or stretch its capacity. Physics and biology impose hard limits.
This was not a problem unique to DMD. Many genetic diseases are caused by large genes that exceed the carrying capacity of AAV vectors, which is why DMD became such an important test case for gene therapy: if scientists could find a way to deliver a functional, truncated dystrophin gene via AAV, the approach could potentially be adapted for other large-gene diseases.
The solution was micro-dystrophin, and it was elegant in its simplicity.
By studying Becker patients with naturally occurring in-frame deletions who remained relatively healthy, researchers identified which portions of the dystrophin protein were essential for function and which were dispensable. They then engineered a dramatically truncated version of the gene, keeping the critical domains that anchor the muscle cell's internal skeleton to its outer membrane while deleting the middle sections. Think of it as preserving the plug and socket ends of an extension cord while removing most of the cable in between. The result was a construct small enough to fit inside an AAV vector while retaining the core structural function of dystrophin.
Sarepta licensed this technology from Nationwide Children's Hospital, paying upfront fees and ongoing royalties for the rights to develop and commercialize the micro-dystrophin construct. In a move that would prove strategically significant, Rodino-Klapac herself eventually joined the company as Executive Vice President and Chief Scientific Officer, bringing the science directly in-house. This was not merely a talent acquisition; it was a transfer of deep institutional knowledge about the gene therapy construct, the manufacturing process, and the clinical development strategy that no amount of licensing could fully capture. The resulting product, designated SRP-9001 and later named Elevidys, used an AAVrh74 capsid, a viral shell selected for its ability to efficiently deliver genes to both skeletal and cardiac muscle.
The early clinical results generated enormous excitement. In Phase 1/2a trials, treated patients showed micro-dystrophin expression in over seventy-six percent of muscle fibers, with intensity reaching roughly three-quarters of normal levels. To put this in perspective, eteplirsen, the exon-skipping drug that had generated so much controversy, produced dystrophin at less than one percent of normal levels. Gene therapy was producing seventy-five times more protein. Creatine kinase, a blood marker of muscle damage that is chronically elevated in DMD patients because their muscle cells are constantly breaking down, dropped by over eighty-seven percent within sixty days of treatment. This was a signal that the muscle membrane was being stabilized, that the fundamental pathology of the disease was being addressed at its source. These were dramatically better numbers than anything the exon-skipping drugs had produced, and they generated a wave of excitement through the DMD research community and among families who had been watching the gene therapy field for years.
On December 23, 2019, Sarepta announced the deal that validated the entire gene therapy bet. Roche, the Swiss pharmaceutical giant, paid $750 million in cash and made a $400 million equity investment in Sarepta, totaling $1.15 billion upfront, for exclusive commercial rights to SRP-9001 outside the United States. The deal also included up to $1.7 billion in regulatory and sales milestone payments and mid-teens royalties on net sales. Sarepta retained all U.S. rights and shared global development costs equally with Roche.
The significance of the Roche deal went beyond the money, though the money was transformative for a company that had spent most of its existence scrounging for capital. Roche's involvement was a validation signal. One of the world's largest and most sophisticated pharmaceutical companies, with deep expertise in drug development and commercialization, had conducted its own due diligence on the science and concluded that Sarepta's gene therapy was worth billions of dollars. For investors, physicians, and patients, this was the ultimate third-party endorsement.
The deal structure was also telling. By retaining all U.S. rights, Sarepta kept the most valuable commercial market for itself. The United States represents roughly half of the global pharmaceutical market and has the most favorable pricing environment for rare disease drugs. Roche took the harder job of navigating the fragmented regulatory and reimbursement landscapes in Europe, Asia, and Latin America. For Sarepta, this was the best of both worlds: Roche's cash funded the development program, Roche's infrastructure would handle international commercialization, and Sarepta kept the U.S. profit pool.
The pivotal EMBARK Phase 3 trial enrolled 125 ambulatory boys with DMD aged four to seven, randomized two-to-one between Elevidys and placebo. The primary endpoint was the change from baseline on the North Star Ambulatory Assessment, or NSAA, a validated seventeen-item functional scale measuring motor abilities like standing from a chair, walking, running, and climbing steps. Each item is scored zero, one, or two, with a maximum total score of thirty-four. Even a single-point change on the NSAA represents a meaningful functional milestone, like the difference between being able to climb a step independently and needing assistance.
The study was designed as a two-part crossover, meaning that after the initial treatment period, placebo patients would also receive Elevidys, allowing long-term follow-up of all participants. This crossover design was both scientifically elegant and ethically necessary: in a fatal childhood disease, no ethics board would approve permanently withholding a potentially beneficial treatment from the control group.
Before the pivotal trial results arrived, Sarepta achieved a major regulatory milestone. On June 22, 2023, the FDA granted accelerated approval to Elevidys for ambulatory DMD patients aged four through five, making it the first gene therapy ever approved for Duchenne. An FDA advisory committee had voted narrowly in favor, eight to six, just weeks before.
The approval was based on micro-dystrophin expression as a surrogate endpoint, using the same regulatory framework that had worked for eteplirsen seven years earlier. The circle was now complete: the controversial precedent that Sarepta had established with eteplirsen was now being used to approve a far more ambitious treatment.
Sarepta priced Elevidys at $3.2 million per dose, making it one of the most expensive drugs in the world at the time of its launch. To put that number in context, the average American household earns roughly seventy thousand dollars per year, meaning a single dose of Elevidys cost the equivalent of approximately forty-five years of median household income. CEO Doug Ingram described the price as "conservative," noting that internal modeling supported a price of five to thirteen million dollars based on quality-adjusted life year calculations.
The value argument rested on the one-time nature of the treatment: unlike exon-skipping drugs requiring lifelong weekly infusions, Elevidys was a single administration intended to provide lasting benefit. Consider a DMD patient receiving Exondys 51 at three hundred thousand dollars per year from age five through twenty-five. That is six million dollars in cumulative drug costs, nearly twice the one-time cost of Elevidys. If the gene therapy worked as hoped, the lifetime cost of treating a DMD patient could actually decrease despite the sticker shock of the initial treatment.
But the Institute for Clinical and Economic Review, the independent drug pricing watchdog, questioned whether the clinical evidence justified any price, let alone $3.2 million. The debate highlighted a fundamental challenge in gene therapy economics: how do you price a treatment when you do not yet know how long it will last?
VIII. The Post-Approval Reality Check (2023-2026)
The story of Elevidys after approval is a case study in the gap between scientific promise and commercial reality, with a safety crisis that shook the entire gene therapy field.
The initial commercial launch was, by any measure, the most successful gene therapy debut in history. For context, Novartis's Zolgensma, a gene therapy for spinal muscular atrophy priced at $2.1 million, was previously considered the gold standard for gene therapy launches, generating approximately $345 million in its first full year. Elevidys obliterated that benchmark. It generated $134 million in revenue in Q1 2024 alone, rising to $384 million in Q4 2024 as the label expanded. In June 2024, the FDA took the unusual step of granting traditional, meaning full, approval for ambulatory patients aged four and above based on the totality of the evidence, while simultaneously granting accelerated approval for non-ambulatory patients. This was remarkable because the EMBARK trial had technically missed its primary endpoint. The fifty-two-week difference between Elevidys and placebo on the North Star Ambulatory Assessment was not statistically significant, with a p-value of 0.24. But the secondary endpoints were significant, including time to rise and the ten-meter walk/run test, and the FDA concluded that the overall evidence package demonstrated clinically meaningful benefit.
The decision was controversial. Critics including ICER's chief medical officer asked a pointed question: if the drug truly worked as well as Sarepta claimed, why had the primary outcome measure failed to show it?
Sarepta's response was that the NSAA is an inherently noisy endpoint in young children, subject to variability from factors as mundane as the child's mood, fatigue, or willingness to cooperate on the day of testing. The secondary endpoints that did achieve significance, like timed rise and the ten-meter walk/run, were more objective and less subject to behavioral variability. The FDA appeared to agree, but the statistical community remained divided.
But for the moment, the commercial machine was running. Elevidys was being infused in patients across the country, and full-year 2024 net product revenue reached $821 million. Combined with nearly a billion dollars from the PMO exon-skipping franchise, Sarepta's total net product revenue hit approximately $1.9 billion, and the company achieved GAAP profitability for the first time in Q4 2024.
Then came 2025, and the narrative collapsed. What followed was the most brutal year in Sarepta's history, and it came just as the company seemed to have finally achieved escape velocity.
The year had started promisingly. Q1 2025 Elevidys revenue came in at $375 million, driven by continued uptake among both ambulatory and non-ambulatory patients. The exon-skipping franchise remained stable at roughly a billion dollars annually. But in March 2025, Sarepta disclosed that a non-ambulatory DMD patient had died of acute liver failure following Elevidys treatment. The stock dropped roughly twenty-five percent in a single day. In June, a second non-ambulatory patient died. A third death followed in July, this time a fifty-one-year-old patient in a clinical trial of SRP-9004, a related gene therapy for limb-girdle muscular dystrophy that used the same AAVrh74 viral vector. The pattern was unmistakable: something about the AAVrh74 vector was causing fatal liver injury in certain patient populations.
The FDA's response was swift and severe. In July 2025, the agency requested that Sarepta suspend all Elevidys shipments, placed every Sarepta clinical trial using the AAVrh74 vector on full clinical hold, and revoked the platform technology designation that had given Sarepta's gene therapy programs expedited review. Sarepta initially resisted the full suspension, then agreed to a voluntary pause on all shipments.
The fallout reshaped the company almost overnight. The market capitalization, which had been above ten billion dollars just months earlier, cratered. Investor confidence evaporated. In July 2025, Sarepta laid off approximately five hundred employees, thirty-six percent of its workforce, and announced four hundred million dollars in annual cost reductions. All limb-girdle muscular dystrophy gene therapy programs except SRP-9003 were discontinued. The FDA ultimately allowed Elevidys to resume for ambulatory patients only, with a new boxed warning, the strongest safety label the FDA can require, for risk of acute serious liver injury and liver failure. The non-ambulatory indication was removed from the label entirely.
The regulatory environment itself had become hostile. Adding to the turbulence, May 2025 had brought the appointment of Dr. Vinay Prasad as head of the FDA's Center for Biologics Evaluation and Research. Prasad was a prominent academic physician who had spent years publicly criticizing Sarepta's therapies and the FDA's accelerated approval process, calling eteplirsen one of the worst FDA decisions in recent history. His appointment sent Sarepta's stock down twenty-six percent in a single day. Prasad's tenure at the FDA was itself chaotic. He resigned in late July amid political backlash from conservative media figures who attacked his handling of Sarepta, was temporarily replaced, then returned in August following a White House review. The episode illustrated how deeply the Sarepta saga had become entangled with broader political debates about FDA regulation.
Meanwhile, the exon-skipping franchise faced its own reckoning. In November 2025, Sarepta announced that the ESSENCE confirmatory trial for Vyondys 53 and Amondys 45 had failed to achieve statistical significance on its primary endpoint, a measure of stair-climbing speed at ninety-six weeks. Sarepta noted that forty-three percent of trial participants had experienced COVID-19-related interruptions that caused missed doses, and that subgroup analyses excluding these patients showed clinically meaningful results. But the headline was damaging: the confirmatory trial had failed, and the question of whether these billion-dollar drugs actually worked remained officially unanswered.
Internationally, the European Medicines Agency's committee issued a negative opinion on Elevidys for the EU market in July 2025, citing insufficient evidence of efficacy. Roche, Sarepta's partner for markets outside the United States, indicated it would continue working with the EMA but the path to European approval remained unclear.
By the end of 2025, the stock had fallen roughly eighty-two percent for the year. The market capitalization that had once exceeded fifteen billion dollars sat below two billion. Elevidys revenue for full-year 2025 was $899 million, essentially flat with 2024 despite a strong first half, as the second-half collapse in non-ambulatory prescriptions erased the earlier gains. The 2026 revenue guidance of $1.2 to $1.4 billion in net product revenue reflected the harsh new reality of an ambulatory-only label.
But the clinical data continued to mature. Three-year EMBARK data presented in January 2026 showed that treated patients maintained motor function above their baseline scores while matched untreated controls showed the expected age-related decline. The treated group demonstrated a seventy-three percent slowing of disease progression on timed rise and seventy percent slowing on the ten-meter walk/run. These were genuinely encouraging numbers, suggesting that for ambulatory patients, the benefit was real and durable. Whether they would be sufficient to rebuild commercial momentum and investor confidence remained to be seen.
In November 2024, anticipating the need to diversify beyond gene therapy, Sarepta had struck a major deal with Arrowhead Pharmaceuticals, licensing seven siRNA programs for $825 million upfront, consisting of $500 million in cash and $325 million in equity, plus up to $10 billion in potential milestone payments. The most significant asset was ARO-DM1, now designated SRP-1003, a treatment for myotonic dystrophy type 1. siRNA, or small interfering RNA, works differently from Sarepta's existing PMO drugs. While PMOs skip over damaged exons to produce a shortened protein, siRNA silences specific genes entirely, blocking the production of toxic or harmful proteins. For myotonic dystrophy, which is caused by a different kind of genetic defect, a repeating DNA sequence that produces a toxic RNA molecule, gene silencing rather than gene restoration is the right therapeutic approach.
This deal signaled a strategic pivot toward chronically-dosed RNA therapeutics for neuromuscular diseases, complementing rather than replacing the gene therapy platform. The Arrowhead portfolio also included preclinical programs for facioscapulohumeral muscular dystrophy and idiopathic pulmonary fibrosis, diversifying Sarepta beyond the DMD focus that had defined it for over a decade. SRP-1003 was already in a Phase 1/2 dose-escalation study, with multiple cohorts completed and data expected in 2026. A second clinical asset, SRP-1005 targeting Huntington's disease, received clinical trial approval in January 2026 and is expected to begin first-in-human dosing in the second quarter of 2026.
Financially, Sarepta ended 2025 with approximately $954 million in cash and investments, against $829 million in long-term debt. In December 2025, the company restructured its near-term debt obligations by exchanging convertible notes maturing in 2027 for new notes maturing in 2030, removing an imminent refinancing pressure. Management projects GAAP profitability and positive cash flow in 2026 despite the lower revenue guidance, driven by the four hundred million dollars in annual cost savings from the July restructuring.
Then, in February 2026, came a deeply personal coda. Doug Ingram announced his retirement, effective by year-end 2026 or upon the appointment of a successor. The reason was simultaneously shocking and poignant: his wife and son had been diagnosed with myotonic dystrophy, the very disease that Sarepta had licensed siRNA treatments for through the Arrowhead deal. The man who had spent nine years fighting for Duchenne families was now, by a cruel twist of fate, a rare disease family himself.
IX. Inflection Points That Changed Everything
Every company's history can be distilled to a handful of moments where the trajectory fundamentally changed. For Sarepta, four decisions stand out as genuinely existential, and each was anything but obvious at the time.
The first was the 2012 strategic refocus and rebranding. When Chris Garabedian decided to shut down the biodefense programs and bet everything on DMD, the company had no approved drugs, no meaningful revenue, and no guarantee that the FDA would ever accept its exon-skipping technology. The prevailing wisdom in biotech is to diversify risk across multiple programs. Garabedian did the opposite, concentrating every resource on a single disease. If the science had failed or the FDA had maintained its skepticism, there was no Plan B. But the focus created clarity, attracted the attention of patient advocacy groups, and built the credibility with the DMD community that would prove essential in every subsequent regulatory battle.
The second was the 2016 eteplirsen approval. This was not merely a drug approval; it was a regulatory precedent. By demonstrating that organized patient advocacy, Congressional pressure, and a sympathetic internal champion at the FDA could overcome negative advisory committee votes and skeptical scientific reviewers, Sarepta created a template that it and other rare disease companies would use repeatedly. The approval also validated dystrophin as a surrogate endpoint, establishing the regulatory pathway that would later enable golodirsen, casimersen, and ultimately Elevidys. Critics argue that this precedent weakened FDA standards; supporters argue it saved lives that would have been lost to bureaucratic caution. Both sides have legitimate points.
The third was the 2019 Roche partnership. Before the deal, Sarepta was developing gene therapy on its own, burning cash at an alarming rate, with no guarantee of success. The $1.15 billion in upfront payments transformed the company's financial position and its strategic options. But the deal also created a powerful signaling effect. When Roche, with its global infrastructure and deep scientific expertise, bet over a billion dollars on Sarepta's gene therapy, it told the market that the technology was real. Every subsequent investor conversation, every FDA interaction, every patient discussion was colored by the fact that one of the world's largest drug companies had validated Sarepta's vision.
The fourth was the June 2023 Elevidys approval, which marked the transition from RNA-based treatments that produced modest amounts of a truncated protein to a gene therapy that delivered meaningful dystrophin expression throughout the body. For the first time, the DMD community could contemplate not just slowing the disease but potentially altering its fundamental trajectory. The commercial implications were enormous: a $3.2 million one-time treatment for a disease that affects thousands of patients worldwide represented a multi-billion dollar opportunity.
A fifth inflection point, one that is still unfolding, was the 2025 safety crisis. The patient deaths and subsequent label restriction represented the most severe setback in the company's modern history, arguably more existential than the 2016 advisory committee rejection because it involved actual patient harm rather than a regulatory opinion. The crisis forced Sarepta to diversify its platform through the Arrowhead siRNA deal, to restructure its cost base, and to confront the possibility that its AAVrh74 gene therapy vector has fundamental safety limitations in certain patient populations. How the company navigates this period will determine whether the 2025 crisis becomes a temporary setback or a permanent inflection toward decline.
Each of these moments was a bet with asymmetric consequences. If any had gone differently, the company might not exist today, or it might be a fraction of its current size. The Sarepta story is a reminder that in biotech, the margin between success and failure is often a single decision, a single vote, or a single data readout.
X. The Playbook: Business and Scientific Lessons
Sarepta's journey offers a set of lessons that are relevant far beyond rare disease biotech, though some of them are uncomfortable to discuss.
The first and most distinctive lesson is that patient advocacy became an integral part of the business strategy. This is not a criticism; it is a description of how the company operated and why it succeeded when others failed. From the eteplirsen approval onward, Sarepta cultivated deep relationships with families, advocacy organizations, and treatment centers. These relationships served multiple functions: they helped identify and recruit clinical trial participants, they generated political pressure during regulatory reviews, they created a constituency that lobbied insurance companies for reimbursement, and they provided a moral narrative that made Sarepta's cause compelling to investors, media, and policymakers.
The ethics of this approach are genuinely complex, and they deserve more discussion than they typically receive in business analyses. Patient advocacy is a democratic good: families affected by devastating diseases have every right to organize, lobby, and demand that their voices be heard in regulatory decisions that affect their lives. But when a company's commercial success depends on those same advocacy groups pressuring regulators to approve products with uncertain evidence, the line between empowerment and exploitation can become blurry. Sarepta's leadership would argue, with some justification, that the company never asked families to advocate for anything that Sarepta did not genuinely believe was in patients' interest. Critics would counter that a $3.2 million drug approval based on a trial that missed its primary endpoint suggests the system is not working as intended.
The second lesson is about regulatory innovation. Sarepta did not merely work within the existing FDA framework; it actively pushed the boundaries of what the accelerated approval pathway could accommodate. The acceptance of dystrophin as a surrogate endpoint, the approval despite negative advisory committee votes, the full approval of Elevidys despite a missed primary endpoint, these were not inevitable outcomes of the regulatory process. They were the result of strategic engagement, legal maneuvering, political pressure, and, to some degree, the FDA's own institutional discomfort with denying treatment to dying children.
The third lesson concerns the power and peril of platform strategy. Sarepta's exon-skipping franchise demonstrated the power of a platform approach in rare diseases: one underlying technology generating multiple products for different genetic subsets of a single disease. The gene therapy program extended this further, potentially offering a mutation-agnostic solution that could treat all DMD patients regardless of their specific genetic defect. But the platform concept also meant that when the AAVrh74 vector encountered safety problems, the damage cascaded across multiple programs simultaneously.
The fourth lesson is about biotech survival. Sarepta's path from penny stock to fifteen billion dollar market cap was not a straight line. It involved reverse stock splits, desperate pivots, government contracts taken for survival rather than strategy, and repeated brushes with bankruptcy. For founders and early-stage biotech investors, the Sarepta story is both inspiring and cautionary: inspiring because it shows that breakthrough science can eventually prevail, cautionary because it took thirty-six years and required multiple near-miracles to get there.
The fifth lesson concerns the art of partnership timing. Sarepta's decision to bring in Roche in 2019, rather than earlier or later, proved crucial. Earlier, and the company would have given away too much of the gene therapy upside before the clinical data had matured. Later, and it might have lacked the financial resources to fund the pivotal trials. The $1.15 billion in upfront payments gave Sarepta the war chest it needed for the EMBARK trial, the manufacturing buildout, and the commercial launch, while retaining full U.S. rights, which is where the overwhelming majority of the value resided.
The sixth lesson is about manufacturing as competitive advantage, and also as competitive risk. Gene therapy manufacturing is extraordinarily difficult, far more so than manufacturing traditional drugs or even biologic antibodies. AAV vectors are produced in mammalian cells through a process that yields mostly empty capsids, viral shells without the therapeutic gene payload. Imagine running a factory where for every ten units you produce, seven or eight are defective and must be discarded. Purification requires separating full capsids from empty ones, a technically demanding process that affects both yield and purity. Batch-to-batch variability is high, and the cost of goods for a single patient dose can reach hundreds of thousands of dollars.
Sarepta invested heavily in internal manufacturing capacity, building an eighty-thousand-square-foot Gene Therapy Center of Excellence in Columbus, Ohio, a thirty-eight-acre campus in Andover, Massachusetts, and additional facilities in Cambridge and Burlington. The company was transitioning from adherent cell culture, where cells grow on flat surfaces, to suspension culture in large bioreactors, a shift that would dramatically increase production capacity. The goal was roughly sixteen hundred doses annually, enough to treat most newly diagnosed DMD patients in the United States each year.
This manufacturing infrastructure, while expensive, was intended to be a competitive moat. Few other companies possessed comparable AAV manufacturing capabilities, and building them from scratch takes years. But the 2025 safety crisis and subsequent restructuring left much of that capacity underutilized, turning what was supposed to be a strength into a financial burden. The irony is particularly sharp: Sarepta spent hundreds of millions building manufacturing capacity for a product whose addressable market was suddenly cut in half by a label restriction.
XI. Power and Competitive Analysis
Assessing Sarepta through Hamilton Helmer's Seven Powers framework reveals a company with genuine but fragile competitive advantages.
The most significant power is Cornered Resource. In rare disease, the resource that matters most is not intellectual property or manufacturing capacity; it is relationships. Sarepta's first-mover access to DMD patients, its accumulated clinical data from over a decade of trials, its relationships with the major neuromuscular disease treatment centers, and its deep ties to advocacy organizations like Parent Project Muscular Dystrophy and CureDuchenne represent assets that competitors cannot easily replicate. When a family receives a DMD diagnosis, the advocacy groups they turn to for guidance often point toward Sarepta's treatments. The physician specialists who treat DMD have years of experience with Sarepta's protocols. These relationships were built through years of genuine engagement, regulatory battles fought together, and shared emotional investment in patient outcomes. A new entrant cannot buy this kind of trust.
Process Power is the second significant advantage. The expertise in exon-skipping chemistry, AAV vector design, rare disease regulatory navigation, and gene therapy manufacturing represents deep organizational knowledge accumulated over decades. Sarepta knows how to design clinical trials for tiny patient populations, how to engage with the FDA on accelerated approval pathways, and how to manage the political dimensions of rare disease drug development. This institutional knowledge is genuinely difficult to replicate.
Switching Costs provide a third layer of advantage, particularly for gene therapy. Once a patient receives Elevidys, the decision is essentially irreversible. The body develops antibodies against the AAVrh74 viral vector, making retreatment with any AAVrh74-based therapy impossible. This means a patient treated with Elevidys cannot switch to a competing gene therapy that uses the same capsid. Physicians who have developed expertise in Sarepta's treatment protocols are also reluctant to change, and the emotional investment families make when choosing a treatment for their child creates psychological switching costs that are as real as the biological ones.
Branding is meaningful within the DMD community. Sarepta is widely seen as the company that "never gave up" on Duchenne, the company that fought the FDA for approval, that lobbied alongside families, that delivered the first gene therapy. This brand equity is genuine and valuable, though it has been damaged by the 2025 safety crisis.
Counter-Positioning originally favored Sarepta. The company challenged FDA orthodoxy on approval standards, positioning itself as patient-first against traditional risk-averse pharmaceutical thinking. But this power diminishes as competitors adopt similar strategies and as the safety events of 2025 validate the concerns of those who argued for more cautious approval standards.
Network Economies are largely absent. Rare disease therapeutics do not benefit from network effects in the traditional sense. Each patient's treatment is independent; having more patients on therapy does not make the therapy more valuable for any individual patient. The only indirect network effect is in clinical data: more treated patients generate more real-world evidence, which can support label expansions and reimbursement negotiations. But this is a weak form of network economy compared to platforms like social media or marketplaces.
Scale Economies exist in manufacturing, where the fixed costs of AAV production facilities can be spread over multiple products, but these advantages are currently diminished by excess capacity and the narrowed label. If Sarepta can successfully develop gene therapies for limb-girdle muscular dystrophy and other conditions using the same AAVrh74 vector and manufacturing infrastructure, scale economies become much more relevant. But the clinical hold on all AAVrh74 programs makes this scenario uncertain.
Turning to Porter's Five Forces, the competitive landscape is more nuanced than Sarepta's historical dominance might suggest, and the dynamics have shifted significantly since the 2025 safety crisis.
The threat of new entrants is moderate to high. The barriers to entry in DMD are substantial: regulatory expertise, clinical trial infrastructure, manufacturing capability, and patient relationships all take years to build. But large pharmaceutical companies can accelerate their entry through acquisition or licensing, as Roche did with Sarepta. More importantly, the gene therapy field is maturing rapidly, and several well-funded competitors are advancing alternatives.
Solid Biosciences is developing SGT-003, a gene therapy using a different AAV capsid that can treat patients who have developed immunity to AAVrh74. Early Phase 1/2 data showed micro-dystrophin expression averaging one hundred ten percent of normal, an extraordinary number that suggests a potentially more potent therapy. REGENXBIO is advancing RGX-202, an AAV8-based micro-dystrophin gene therapy with its own encouraging Phase 1/2 data and plans to submit for approval in mid-2026. Both use different viral vectors, meaning they could treat patients who are ineligible for Elevidys due to pre-existing antibodies against AAVrh74.
In the exon-skipping space, next-generation competitors are emerging with potentially superior technology. Dyne Therapeutics has shown dystrophin expression of nearly nine percent with its exon 51 skipping drug, more than tenfold higher than what Exondys 51 produced. Avidity Biosciences has demonstrated dystrophin production up to twenty-five percent of normal, among the best exon-skipping data ever recorded. If these results hold up in larger trials, Sarepta's first-generation PMO drugs could face serious competitive pressure.
The bargaining power of buyers is moderate but increasing. This is the force that investors should watch most carefully in the near term. Insurance companies and government payers are pushing back on multi-million dollar gene therapy prices, and the political environment around drug pricing is growing more hostile. The $3.2 million price tag for Elevidys, while defensible on a per-patient cost-effectiveness basis, is politically vulnerable. If outcomes-based contracts become standard, Sarepta's revenue could become tied to long-term efficacy data that is still maturing.
The bargaining power of suppliers is moderate. AAV vector manufacturing requires specialized equipment, raw materials, and expertise that are in limited supply across the industry. Sarepta's dependence on the Nationwide Children's Hospital intellectual property for its gene therapy construct is a specific supplier risk, though the licensing relationship appears stable.
The threat of substitutes is moderate to high and growing. This is perhaps the most underappreciated risk in the Sarepta story. Beyond competing gene therapies, CRISPR-based gene editing represents a longer-term but potentially transformative alternative. If CRISPR can eventually correct DMD mutations at the DNA level, rather than delivering a miniaturized replacement gene via a viral vector, the entire gene therapy approach could become obsolete. No CRISPR DMD therapy has reached late-stage clinical trials, but the technology is advancing rapidly.
Industry rivalry is moderate but intensifying. The DMD therapeutic landscape has more participants today than at any point in history. Pfizer's exit from DMD gene therapy in mid-2024, after its fordadistrogene movaparvovec failed its Phase 3 trial and caused one patient death, temporarily reduced competition in the gene therapy lane. But the vacuum did not last. As described above, Solid Biosciences and REGENXBIO are both well-funded and advancing with differentiated AAV capsids that could reach patient segments Sarepta cannot. The fact that both competitors use different viral vectors means they are not just fighting for market share; they are expanding the treatable population in ways that could marginalize Elevidys.
In the exon-skipping space, the competitive threat is arguably even more acute. If Dyne's and Avidity's next-generation drugs win approval with dystrophin expression ten to twenty-five times higher than Sarepta's first-generation PMOs, they could render Exondys 51, Vyondys 53, and Amondys 45 obsolete, threatening a billion-dollar revenue stream that currently funds the rest of Sarepta's operations. The confirmatory trial failure for the PMO drugs adds regulatory vulnerability on top of the competitive threat.
The overall assessment is that Sarepta operates in a structurally attractive rare disease niche with meaningful competitive advantages, but those advantages are less durable than they appeared two years ago. The safety crisis damaged the brand, narrowed the label, and created an opening for competitors with different viral vectors. The confirmatory trial failures for the exon-skipping franchise remain a regulatory overhang. And the CEO transition adds management uncertainty at a moment when strategic clarity is essential.
XII. Bull vs. Bear Case
The current stock price, hovering around seventeen dollars per share in early March 2026, represents a market capitalization of roughly $1.75 billion. For a company generating over two billion dollars in annual revenue, that valuation implies severe skepticism about the sustainability of the revenue base. The bull case for Sarepta rests on the durability of the Elevidys data and the company's unmatched position in DMD.
Three-year EMBARK data show that treated ambulatory patients are maintaining motor function while untreated patients decline, with seventy percent to seventy-three percent slowing of disease progression on key functional measures. If this trend continues, Elevidys could fundamentally change the trajectory of DMD for ambulatory patients, justifying both its price and its position as the standard of care. The majority of eligible ambulatory patients have not yet been treated, representing a substantial multi-year commercial opportunity even within the restricted label. The PMO exon-skipping franchise continues to generate nearly a billion dollars in annual revenue, providing a stable cash flow base while the gene therapy business recovers. The Arrowhead siRNA deal diversifies the pipeline into myotonic dystrophy, Huntington's disease, and other neuromuscular conditions, creating optionality beyond DMD. And if the ENDEAVOR Cohort 8 study of an enhanced immunosuppressive regimen demonstrates safety in non-ambulatory patients, the recapture of that lost indication could be transformative.
The Sarepta of March 2026 also has a path to non-ambulatory market recapture. The company received FDA approval to initiate ENDEAVOR Cohort 8, a study evaluating an enhanced sirolimus immunosuppressive regimen before and after Elevidys infusion in non-ambulatory patients. The hypothesis is that a more aggressive approach to suppressing the immune system before treatment could prevent the fatal liver reactions that occurred in 2025. If the data are positive, a label change restoring the non-ambulatory indication could come as early as 2027, which would dramatically expand the commercial opportunity.
The bear case is equally compelling and grounded in recent events.
The safety crisis was not an abstract risk; three patients died. The non-ambulatory market, which represented a significant portion of Elevidys's commercial potential, has been lost and may never be recovered. Gene therapy durability remains unknown. AAV-delivered micro-dystrophin sits as an extrachromosomal element in muscle cells, meaning it does not integrate into the cell's DNA. While mature muscle fibers do not divide, the satellite cells responsible for muscle repair and regeneration do divide, and each division dilutes the therapeutic gene. If the benefit fades after five or ten years, retreatment is currently impossible because the body's immune system has already learned to recognize and destroy the AAVrh74 vector.
The confirmatory trials for the exon-skipping drugs have failed, and the FDA could eventually withdraw those approvals, eliminating a billion-dollar revenue stream. Historically, every drug that has failed its confirmatory trial after accelerated approval has been withdrawn. Sarepta has argued that real-world evidence and subgroup analyses support continued approval, but the precedent is not in the company's favor.
Competitors with different AAV capsids could offer gene therapies that are both safer and effective in patient populations that Elevidys cannot reach. The $3.2 million price tag is politically vulnerable, and the smaller the eligible patient population becomes, the more pressure each individual sale faces from payers. The CEO transition introduces management uncertainty at a critical juncture.
For investors tracking Sarepta's ongoing performance, two metrics matter most.
The first is quarterly Elevidys infusion revenue, which serves as the real-time indicator of commercial recovery following the label restriction. This number captures patient identification, physician adoption, payer coverage, and manufacturing execution in a single figure. A sustained recovery toward pre-crisis run rates would signal that the ambulatory market alone can support meaningful growth. A continued decline would suggest that the safety stigma is suppressing demand beyond what the label restriction alone explains.
The second is long-term Elevidys durability data, specifically the EMBARK results at four years and beyond. Gene therapy is predicated on the idea that a single treatment produces lasting benefit. If treated patients continue to diverge favorably from untreated controls on functional measures, the clinical case for the drug strengthens with time, supporting both broader adoption and pricing sustainability. But if the benefit plateaus or reverses, the entire commercial thesis unravels: a $3.2 million treatment that lasts only three to five years would be difficult to justify when lifelong exon-skipping drugs, while less effective per dose, produce continuous treatment.
Everything else, pipeline progress, competitive dynamics, reimbursement trends, flows from these two fundamental indicators. Watch them closely.
XIII. Epilogue: What Comes Next
The Sarepta story is, in many ways, the story of gene therapy itself. A technology that was dismissed as science fiction for decades, that faced repeated failures and setbacks, that required unprecedented manufacturing innovations and regulatory compromises, has now produced a real treatment for a real disease that is changing real lives. The question is whether the current setbacks represent a permanent limitation or a temporary growing pain.
The rare disease drug development model that Sarepta pioneered, built on patient advocacy, accelerated approval pathways, and surrogate endpoints, is now being tested in the most consequential way possible: with actual patient outcomes. If the three-year EMBARK data hold up and extend, and if the ambulatory safety profile remains clean, the model will be vindicated. If the data plateau or new safety signals emerge, the entire framework of accelerated approval based on surrogate biomarkers will face renewed and justified scrutiny. Either way, the implications extend far beyond DMD to every rare disease program working through the FDA.
For the DMD community, the emotional trajectory has been particularly cruel. Families spent decades being told their children's disease was untreatable, then experienced the exhilaration of eteplirsen's approval, then watched as the clinical benefit remained contested, then celebrated the gene therapy breakthrough, then endured the horror of patient deaths and label restrictions. The emotional whiplash mirrors the stock chart, and behind every data point is a family making the most consequential medical decision of their lives with incomplete information.
The broader implications of the Sarepta story extend well beyond the company and Duchenne. The patient advocacy model that Sarepta helped pioneer has been adopted by rare disease communities across dozens of conditions. The regulatory precedents established through eteplirsen, for better or worse, have shaped how the FDA evaluates drugs for small patient populations with unmet needs. The manufacturing capabilities that Sarepta and others have built are laying the groundwork for gene therapies in other diseases. And the pricing and reimbursement debates triggered by Elevidys are forcing societies to confront fundamental questions about how much a child's life is worth and who should pay for the answer.
The competitive landscape over the next several years will determine whether Sarepta maintains its dominant position or becomes a cautionary tale about first-mover disadvantage in a rapidly evolving field. In technology, first movers often win because network effects compound their advantage. In biotech, first movers sometimes lose because their early products become obsolete as the science advances. Solid Biosciences and REGENXBIO are advancing gene therapies with different viral vectors that could be safer and more broadly applicable. Next-generation exon-skipping drugs from Dyne and Avidity may significantly outperform Sarepta's first-generation PMOs. CRISPR-based approaches, while still early, represent the ultimate long-term competitive threat: if you can fix the gene itself rather than delivering a miniaturized replacement, the entire current therapeutic paradigm becomes obsolete.
For Sarepta specifically, the path forward requires navigating multiple simultaneous challenges. Rebuilding commercial momentum for Elevidys in the ambulatory population. Resolving the clinical holds on the LGMD gene therapy programs. Advancing the siRNA pipeline acquired from Arrowhead into late-stage development. Managing the CEO succession without losing strategic coherence. And above all, demonstrating that gene therapy durability holds up over time. The company's 2026 guidance of $1.2 to $1.4 billion in net product revenue represents a significant step down from 2025's nearly $1.9 billion, and the path back to growth depends on execution in every one of these areas.
Doug Ingram's final act as CEO carried a poignancy that no corporate communications team could have scripted. The man who spent nine years championing treatments for other people's children learned that his own family carried a different form of muscular dystrophy. That his company had, just months earlier, licensed a potential treatment for that very disease from Arrowhead Pharmaceuticals was either an extraordinary coincidence or a testament to Ingram's strategic foresight.
Either way, it collapsed the distance between executive and patient, between business strategy and personal stakes, in a way that captured the essential truth of rare disease drug development: this is not abstract. These are real people, real families, and the consequences of success or failure are measured not in stock prices but in lifetimes. In his retirement announcement, Ingram reportedly told colleagues that he wanted to spend his remaining time with his family in California while they still had the years of relatively normal function ahead of them. The urgency was personal now, not professional.
The ultimate question remains unanswered: can we actually cure Duchenne? The honest answer is that we do not yet know. What we know is that for the first time in history, boys with Duchenne have access to treatments that can slow the disease's relentless progression, and that the science of gene therapy and gene editing continues to advance at a pace that makes genuine cures plausible within the next decade.
Sarepta has been at the center of this progress for longer and through more adversity than any other company. It has survived penny-stock status, near-bankruptcy, regulatory rejection, the most controversial FDA approval in a generation, a transformative big pharma partnership, the first gene therapy approval for the disease, patient deaths, stock collapse, and the impending departure of the leader who steered it through most of that journey. Few companies in any industry have experienced such dramatic swings between triumph and catastrophe in such a compressed timeframe.
Whether Sarepta will be the company that ultimately delivers a cure, or whether it will be remembered as the pioneer that blazed the trail for others, is the question that the next chapter of this remarkable story will answer.
XIV. Further Reading
Sarepta Therapeutics remains one of the most closely watched companies in biotech, and the story continues to develop rapidly. For those who want to go deeper, the following resources provide essential context and primary source material on the company and the Duchenne landscape.
The Boston Globe's investigative series "The Boy Who Would Be Saved" remains the definitive account of the eteplirsen approval controversy, documenting the internal FDA battles, the patient advocacy campaign, and the political pressures that shaped the 2016 decision.
The FDA's publicly available briefing documents for both eteplirsen and Elevidys advisory committee meetings provide unfiltered access to the scientific debates, the clinical data, and the reviewers' concerns that are often sanitized in media coverage.
Sarepta's SEC filings, particularly the 10-K annual reports and proxy statements, offer the most detailed account of the company's strategic evolution, financial trajectory, and risk factors.
Parent Project Muscular Dystrophy and CureDuchenne's public resources provide the patient perspective that is essential for understanding why the advocacy community was so effective and why the stakes felt so personal.
Nature Reviews Drug Discovery has published multiple in-depth articles on exon-skipping and DMD therapeutics that explain the science at a level accessible to non-specialists.
Siddhartha Mukherjee's "The Gene" provides broader context on the genetic medicine revolution that made Sarepta's work possible.
BioCentury and Endpoints News archives contain the most detailed contemporaneous coverage of Sarepta's clinical and regulatory milestones.
The JAMA and New England Journal of Medicine publications on the clinical trial results and the medical community's debates about accelerated approval offer the scientific community's unvarnished assessment of Sarepta's evidence base.
Roche's partnership press releases and investor presentations provide the strategic rationale behind the largest gene therapy licensing deal in history.
And the Congressional hearing transcripts from 2013 through 2016, where DMD families testified before lawmakers about their children's lives and the urgency of drug approval, remain some of the most powerful documents in the history of patient advocacy in action.
 RSS Feed
RSS Feed Spotify
Spotify Apple Podcasts
Apple Podcasts Amazon Music
Amazon Music Audible
Audible YouTube
YouTube