ACADIA Pharmaceuticals: The Story of a CNS Drug Developer's Improbable Journey
I. Introduction & Episode Roadmap
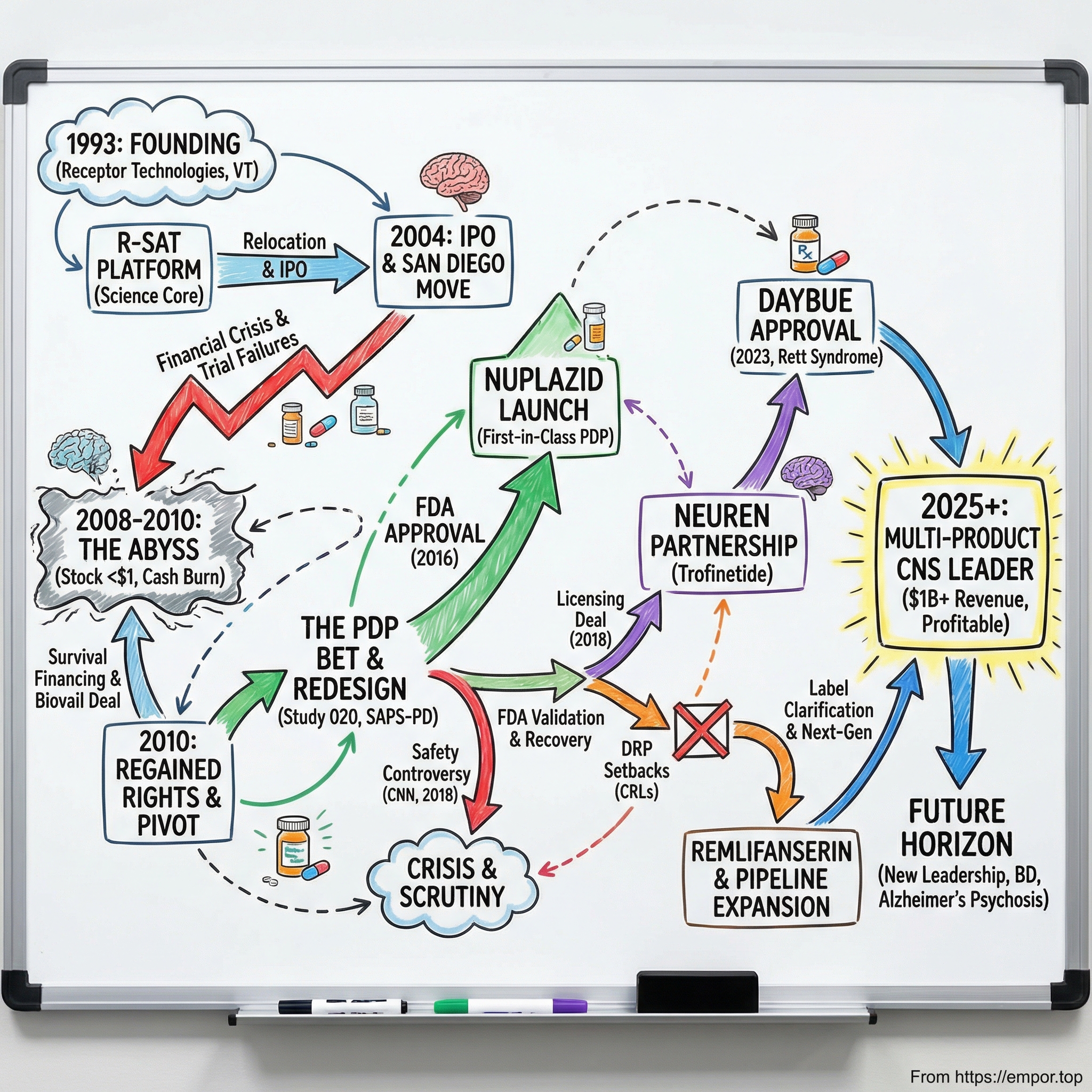
Picture a small pharmaceutical company, barely clinging to life, its stock trading under a dollar, running clinical trials that keep failing, burning through cash it cannot replace. Now picture that same company, three decades later, crossing a billion dollars in annual revenue, with two first-in-disease FDA-approved drugs and a pipeline that could define the next decade of neuropsychiatric medicine. That is the story of ACADIA Pharmaceuticals.
ACADIA is not the kind of company that gets splashy magazine covers. It operates in the unglamorous corridors of central nervous system drug development, a field so littered with failures that most large pharmaceutical companies have abandoned it entirely. And yet, from its founding as "Receptor Technologies" in a small Vermont lab in 1993 to its current position as the dominant player in Parkinson's disease psychosis and the only company with an approved treatment for Rett syndrome, ACADIA has pulled off something remarkable: it survived.
The company's story is a masterclass in the biotech risk-reward paradox. For every Moderna that rockets from obscurity to a hundred-billion-dollar valuation, there are hundreds of biotech companies that burn through investor capital and disappear without ever bringing a product to market. ACADIA nearly joined that list, multiple times. The stock crashed below sixty-five cents during the financial crisis, hitting its all-time low in November 2010. Clinical trials failed repeatedly. The FDA rejected a key expansion — twice. A CNN investigation threatened to pull its only approved drug from the market.
Each time, the company found a way to survive, pivot, and ultimately prevail.
What makes this story worth telling in depth is not just the science or the financials, but the strategic decisions made under extreme uncertainty. How do you choose the right disease to target when your drug has failed in three indications? How do you convince the FDA to approve a medication based on a single positive trial when three previous trials came up negative? How do you weather a media firestorm alleging your drug is killing patients? And how do you transform from a one-product company into a diversified rare disease leader?
This is a story about persistence, scientific conviction, and the uncomfortable truth that in drug development, the line between a billion-dollar blockbuster and a write-off is often razor thin.
Let's start at the beginning.
II. Founding Context & The Neuroscience Gold Rush (1993-2000)
In the early 1990s, the biotechnology industry was riding a wave of optimism fueled by the Human Genome Project and the promise that unlocking the genetic code would revolutionize medicine. Venture capital was flowing into biotech startups at an unprecedented pace, and among the most tantalizing frontiers was the central nervous system. The brain, with its billions of neurons and trillions of synaptic connections, represented the ultimate drug development challenge and the ultimate prize.
Into this heady environment stepped Mark R. Brann, a pharmacologist with a Ph.D. and a conviction that the future of CNS medicine lay in understanding receptor biology. In 1993, Brann founded a company called Receptor Technologies in Winooski, Vermont, a small city just north of Burlington, about as far from the biotech hubs of San Diego and Boston as you could get. The company's founding thesis was deceptively simple: if you could precisely understand how drugs interact with specific receptors in the brain, you could design better, more selective medicines with fewer side effects.
Brann was joined by co-founder Sherman M. Weissman, and together they built a proprietary technology platform called R-SAT, which stood for Receptor Selection and Amplification Technology. This was essentially a high-throughput screening system that allowed researchers to test thousands of chemical compounds against hundreds of cellular receptor targets simultaneously. Think of it as a dating app for molecules and receptors, identifying which ones would make the best matches.
The early funding came from an unlikely source: Danish investors. BankInvest, led by managing director Florian Schönharting, along with several Danish pension funds and private investors, bankrolled the fledgling company. At its peak, ACADIA maintained a Denmark office with fourteen of its roughly fifty employees stationed there, including a medicinal chemistry operation in Copenhagen.
This Scandinavian connection would later play a role in recruiting leadership talent, and it foreshadowed something important about ACADIA's DNA: the company was always willing to look far afield for the right people and the right ideas.
By 1997, the company had outgrown Vermont. The founders made a pivotal decision to relocate all operations and management to San Diego, California, placing themselves in the heart of one of the world's premier biotech ecosystems. San Diego's Torrey Pines Mesa, home to the Salk Institute, Scripps Research, and UC San Diego, had been producing biotech talent and companies since the 1960s. The success of Hybritech, acquired by Eli Lilly for four hundred million dollars in the 1980s, had ignited venture capital investment in the region, and roughly a quarter of San Diego's life science companies could trace their roots to Hybritech alumni. More than fourteen hundred life science companies now operate in the region, along with over fifty research institutions. The ecosystem's deep expertise in neuroscience, combined with a dense network of contract research organizations and potential pharmaceutical partners, made it the natural home for a CNS-focused startup.
The move also brought new leadership. In 2001, Uli Hacksell, a Swedish pharmacologist who had spent his career in medicinal chemistry and neuroscience research at Uppsala University and later in the pharmaceutical industry at Astra, took over as CEO. Hacksell brought a deeply scientific orientation to the role, a data-driven approach rooted in his academic and R&D background that would guide ACADIA through its most challenging years of drug development. Mark Brann stayed on as President and Chief Scientific Officer until 2006, when he departed to launch Abbey Pharmaceuticals, a company focused on substance abuse therapeutics.
The company was renamed ACADIA Pharmaceuticals, and it began building partnerships. Three separate collaborations with Allergan, the eye care and aesthetics giant, provided research funding and validation. Allergan took a 6.3 percent ownership stake, and the partnership focused on drug discovery for chronic pain, glaucoma, and other receptor-mediated conditions. These deals kept the lights on during ACADIA's years of pure R&D spending with zero revenue.
ACADIA went public in May 2004, raising a modest thirty-five million dollars in its IPO, well short of the sixty-four million the company had originally targeted. At the time, it had five drugs in development with two in human trials. The IPO was not a blockbuster, but it gave the company the public currency it would desperately need in the years ahead. Three years later, on the strength of promising early clinical data, ACADIA completed a secondary offering in May 2007 that raised a hundred and two million dollars. The stock peaked around sixteen dollars that October.
Then the world fell apart.
But before we get to the financial crisis, we need to understand what ACADIA was building in its labs, because the science is where this story really begins to get interesting.
III. The Early Pipeline: Promise and Painful Reality (2000-2010)
To understand ACADIA's journey, you need to understand a bit about brain chemistry, and specifically about a molecule called serotonin. Serotonin is one of the brain's most important chemical messengers, influencing everything from mood and sleep to perception and cognition. It acts through a family of receptors, and one of the most important is called the 5-HT2A receptor.
Here is the key insight that drove ACADIA's entire scientific strategy: virtually every classic hallucinogen, LSD, psilocybin, mescaline, produces its mind-altering effects primarily by activating the 5-HT2A receptor. When this receptor is overstimulated, it cranks up cortical excitability and disrupts normal perception, creating hallucinations and psychotic-like experiences. Conversely, nearly all atypical antipsychotic drugs, the medications used to treat schizophrenia and other psychotic disorders, work in part by blocking this same receptor.
ACADIA's scientists, using their R-SAT platform, made a critical discovery in the late 1990s. They profiled a comprehensive library of marketed CNS drugs across hundreds of receptor targets and found that potent "inverse agonist" activity at the 5-HT2A receptor was a common feature of the atypical antipsychotics that worked best in treating psychosis. This was not just academic; it was a potential drug development roadmap.
Now, a quick explanation of terminology that matters here. A regular "antagonist" is like putting a cork in a bottle; it blocks the receptor so nothing can activate it. An "inverse agonist" goes further. Many receptors, including 5-HT2A, have what scientists call "constitutive activity," meaning they are partially switched on even when no signaling molecule is present. An inverse agonist does not just block the receptor; it actively pushes the receptor's activity below baseline, silencing this background chatter. Think of it as not just muting a speaker, but actually pulling the plug.
The ACADIA team screened a hundred and thirty thousand compounds using R-SAT, generating about five hundred initial hits. A hundred of these were characterized as potent 5-HT2A inverse agonists, and chemical optimization produced a lead compound called AC-90179. It was highly selective for the 5-HT2A receptor, nearly a hundred-fold more selective than for related receptors. The problem was that it had terrible oral bioavailability, meaning the body could not absorb it efficiently when taken as a pill. Back to the drawing board.
Further medicinal chemistry work produced a refined molecule: pimavanserin, initially designated ACP-103.
This compound was a breakthrough. It maintained nanomolar potency as a 5-HT2A inverse agonist, had excellent selectivity, and crucially, much greater oral bioavailability. And here was the most important feature: unlike traditional antipsychotics, pimavanserin had no meaningful affinity for dopamine D2 receptors.
Why does that matter? Because the most devastating neurological diseases involving psychosis, particularly Parkinson's disease, are caused by the loss of dopamine neurons. Parkinson's patients take dopamine-replacing medications like levodopa and dopamine agonists to manage their tremors, rigidity, and slowness of movement. Traditional antipsychotics that block dopamine receptors directly counteract these medications, creating an impossible clinical dilemma: treat the hallucinations but worsen the movement disorder, or preserve motor function but leave the patient tormented by visions and delusions.
To appreciate the severity of this problem, consider that before NUPLAZID, the only antipsychotic with any evidence in Parkinson's psychosis was low-dose clozapine, which requires weekly blood monitoring due to the risk of a potentially fatal drop in white blood cell count. It is one of the most burdensome drugs in all of medicine to prescribe and take. The alternative, quetiapine, had never been shown to work in randomized trials for PDP and still carried dopamine-blocking side effects. Physicians were essentially left with no good options. Pimavanserin promised to break this deadlock.
But first, ACADIA had to prove it worked, and the path forward was brutal. In 2007, the company announced positive results from a Phase II co-therapy trial of ACP-103 combined with risperidone in schizophrenia patients. The data looked promising. Then in June 2008, disaster struck: ACP-104, a related compound, failed its Phase IIb trial in schizophrenia. Neither dose showed improved efficacy compared to placebo. The stock cratered.
And then came the financial crisis. ACAD shares plummeted from around sixteen dollars in late 2007 to under one dollar by March 2009, eventually bottoming at sixty-five cents on November 12, 2010. A decline of more than ninety-five percent from peak to trough. The biotech sector was hemorrhaging: over forty-two thousand pharmaceutical industry jobs were cut in just the first four months of 2009. ACADIA was burning cash with no revenue, failed trials behind it, and a stock price that made further equity raises nearly impossible.
To survive, the company secured a Committed Equity Financing Facility with Kingsbridge Capital, which committed to provide up to sixty million dollars over three years through the purchase of newly issued shares. It was dilutive and painful, but it kept ACADIA alive. A fifteen million dollar private placement in January 2011 provided additional runway. The company also entered a partnership with Biovail Laboratories, granting North American rights to pimavanserin in exchange for a thirty million dollar upfront payment and up to three hundred and twenty million in milestone payments. This deal was a lifeline.
But then, just seventeen months later, something remarkable happened. ACADIA and Biovail concluded their collaboration in October 2010, and ACADIA regained all North American rights to pimavanserin. Biovail paid ACADIA a one-time cash payment of eight million seven hundred and fifty thousand dollars to cover the transition. ACADIA had bought back its future, but it was a bet on a drug that had not yet proven itself in the indication that would ultimately matter most.
IV. The Parkinson's Disease Psychosis Bet: Finding the Right Disease
The decision that saved ACADIA Pharmaceuticals was not made in a moment of inspiration. It was made through a process of elimination, after watching promising indications fail one after another, and then noticing something that everyone else had overlooked.
Parkinson's disease psychosis, or PDP, is a condition that most people outside of neurology have never heard of. But for the roughly one million Americans living with Parkinson's disease, psychosis is one of the most feared and devastating complications. Approximately fifty percent of Parkinson's patients will experience hallucinations or delusions at some point during the course of their illness. The hallucinations are most commonly visual: patients see people who are not there, insects crawling on walls, animals in the room. They are often vivid, detailed, and terrifying.
The delusions can be equally destructive. A patient might become convinced their spouse is unfaithful, that family members are stealing from them, or that they are being watched. These symptoms place an enormous burden on caregivers, are a leading cause of nursing home placement, and are associated with significantly increased mortality.
And here was the critical fact that caught ACADIA's attention: as of the late 2000s, there was not a single FDA-approved treatment for Parkinson's disease psychosis. Zero. The standard of care was to prescribe antipsychotics "off-label," medications approved for schizophrenia that had never been studied in Parkinson's patients. These drugs block dopamine receptors, which, as we discussed, directly worsens the motor symptoms of Parkinson's disease. It was medicine's version of robbing Peter to pay Paul.
The clinical trial design challenges were formidable. Measuring psychosis in Parkinson's patients is inherently difficult because the symptoms fluctuate, the patients are elderly and frail, and the placebo response rates in psychiatric trials are notoriously high. Traditional psychosis rating scales, designed for schizophrenia patients, were not well suited to capturing the specific hallucinatory and delusional experiences of PDP patients.
ACADIA initiated a Phase II study of pimavanserin in PDP patients, designated ACP-103-002. This was a four-week, sixty-patient study that showed encouraging signals of efficacy, particularly in reducing persecutory delusions, which achieved statistical significance. But it was not a slam-dunk, and much larger trials would be needed.
The company then embarked on Phase III trials, and this is where the story gets painful again. The first Phase III trial, ACP-103-012, enrolled two hundred and ninety-eight patients internationally and tested ten milligram and forty milligram doses against placebo over six weeks. The trial failed. An unexpectedly large placebo response of forty-two percent, meaning patients getting sugar pills improved dramatically, swamped the drug signal. The forty milligram dose showed promising trends, but nothing that would satisfy the FDA.
A second Phase III trial, ACP-103-014, was stopped early after enrolling only a hundred and twenty-three patients, as the lower doses of ten and twenty milligrams showed only numerical, not statistical, separation from placebo.
Two Phase III failures in the indication that was supposed to save the company.
For most biotech companies, this would be the end. The pipeline would be declared dead, the remaining cash returned to shareholders, and the company wound down. But ACADIA's scientists and leadership believed the drug worked; they just needed to figure out the right way to prove it.
The critical insight was that they needed to redesign everything: the dose, the patient selection, the trial procedures, and most controversially, the measurement scale. For the pivotal third trial, ACP-103-020, ACADIA implemented several key changes. They went with a one-to-one randomization instead of three-arm designs. They introduced a two-week lead-in period of psychosocial therapy before randomization, designed to wash out the placebo response that had torpedoed previous trials. They focused exclusively on the forty milligram dose. And they developed a new nine-item assessment scale called the SAPS-PD, derived from the broader Schedule for the Assessment of Positive Symptoms, specifically selecting the items most relevant to Parkinson's disease psychosis.
This last decision was controversial, and it would shadow the company for years.
The SAPS-PD was created through post-hoc factor analysis of the failed trials, essentially mining the data from Studies 012 and 014 to identify which specific questions best captured the treatment signal. Critics would later argue this was a form of cherry-picking, designing a scale that was optimized to make your drug look good. ACADIA argued it was good science: identifying the right tool for the right disease.
The result was Study ACP-103-020, and it succeeded. Pimavanserin forty milligrams demonstrated a highly significant improvement on the SAPS-PD compared to placebo, with a p-value of 0.0014 and an effect size of 0.50. A thirty-seven percent improvement in psychosis symptoms versus fourteen percent for placebo. The drug also improved clinical global impression scores, caregiver burden, nighttime sleep quality, and daytime wakefulness, all without worsening motor symptoms. In preclinical models using bilateral substantia nigra lesion rats, pimavanserin had reversed psychosis-like behaviors without impairing motor function or blocking the effectiveness of L-DOPA, advantages that quetiapine and clozapine could not match in the same model. The clinical trial data confirmed what the preclinical work had predicted.
It had taken more than fifteen years, multiple failed trials, near-bankruptcy, and a complete rethinking of clinical trial methodology, but ACADIA finally had data that could support an FDA filing. The real question now was whether the FDA would agree.
V. The FDA Approval Drama: Inflection Point #1 (2013-2016)
In April 2013, ACADIA secured an alignment meeting with the FDA, during which the agency agreed that the data from Study 020, combined with supportive data from earlier trials, could form the basis of a New Drug Application. This was a crucial milestone: the FDA was saying, in effect, "we are willing to look at this." Then in September 2014, the agency granted Breakthrough Therapy Designation for pimavanserin in Parkinson's disease psychosis. This designation, created by Congress in 2012, was reserved for drugs that demonstrated "substantial improvement over existing therapies" for serious conditions. Given that there were no existing therapies for PDP, the bar was effectively: does this drug work at all? The Breakthrough designation gave ACADIA more intensive FDA guidance and a potentially faster path to approval.
By September 2015, ACADIA submitted its New Drug Application. The FDA accepted it in October with Priority Review, setting a target action date of May 1, 2016. The filing was based primarily on the single positive Phase III trial, Study 020, supplemented by supportive data from the earlier failed trials and open-label extension studies showing continued safety and tolerability.
The FDA's Psychopharmacologic Drugs Advisory Committee convened on March 29, 2016, and this is where the drama intensified. The committee had to weigh two uncomfortable truths simultaneously. First, the drug showed statistically significant efficacy and represented a genuine first-in-class treatment for an unmet need. Second, the safety data contained troubling signals.
The FDA's medical reviewer, Dr. Paul Anderson, recommended against approval. His analysis concluded that treatment with pimavanserin "more than doubled the risk of death and/or serious adverse events" compared to placebo. This was not a minor footnote; it was a senior FDA scientist saying this drug might be killing people.
The advisory committee heard testimony from Parkinson's disease patients and caregivers, many of whom described the devastating impact of psychosis on their lives. They heard from KOLs who argued that the alternative, off-label use of dopamine-blocking antipsychotics, was far worse. And they grappled with the fundamental question that runs through all drug regulation: how much risk is acceptable when the alternative is no treatment at all?
The vote was twelve to two in favor, a strong recommendation that the benefits of pimavanserin outweighed its risks.
On April 29, 2016, two days ahead of the target action date, the FDA approved NUPLAZID, making pimavanserin the first and only FDA-approved treatment for hallucinations and delusions associated with Parkinson's disease psychosis.
The approval came with a Boxed Warning, the FDA's most serious label warning, about increased risk of death in elderly patients with dementia-related psychosis treated with antipsychotic drugs. Specifically, the label stated that in seventeen placebo-controlled trials of antipsychotics in dementia patients, the death rate was about four and a half percent on drug versus two and a half percent on placebo, roughly double the risk. NUPLAZID was explicitly not approved for treating psychosis in dementia patients without Parkinson's disease. Additional warnings addressed QT prolongation, a heart rhythm concern.
For ACADIA, this was the validation of more than two decades of work. Twenty-three years from founding to first drug approval. The stock surged. Steve Davis, who had joined as CFO and Chief Business Officer in July 2014 and been named interim CEO in March 2015 after Hacksell's retirement, was confirmed as permanent CEO in September 2015. Davis was an unusual choice for a biotech CEO: a CPA and attorney by training, with a J.D. from Vanderbilt, he brought an operational and commercial lens that would prove critical for the next phase. His prior experience at Heron Therapeutics and Ardea Biosciences had given him the playbook for building a commercial organization from scratch. Now he would have to execute it.
The approval came at a time when ACADIA had virtually no commercial infrastructure. It had gone from a company of researchers and regulators to one that needed a sales force, a distribution network, a patient support program, and a marketing strategy, essentially overnight. The company could now begin the transition from a money-losing R&D operation to a commercial-stage pharmaceutical company. But this approval also planted the seeds for the next crisis. The safety questions raised during the advisory committee would not stay buried for long.
VI. Commercial Launch & The Safety Controversy (2016-2018)
NUPLAZID became available for prescription on May 31, 2016, barely a month after FDA approval. ACADIA had been preparing for this moment, hiring approximately a hundred and thirty-five seasoned sales specialists with an average of eight years of CNS sales experience and fifteen years in the pharmaceutical industry. The sales force was divided into two groups: roughly a hundred representatives targeting office-based neurologists and movement disorder specialists, and about fifty in an "institutional" team focused on nursing homes and long-term care facilities.
The commercial strategy reflected the unusual nature of PDP as a market. This was not a primary care drug that would be prescribed by tens of thousands of family doctors. PDP was a specialty condition, diagnosed and treated by a relatively small number of neurologists, psychiatrists, and movement disorder specialists. ACADIA launched NUPLAZIDconnect, a patient support program offering financial assistance and access help, along with a thirty-day free trial for new patients. In 2017, the company debuted its first branded direct-to-consumer advertising campaign to raise awareness that PDP was a real, treatable condition.
The revenue ramp was dramatic. In 2016, the partial launch year, NUPLAZID generated seventeen million three hundred thousand dollars. In 2017, its first full year, revenue leapt to a hundred and twenty-five million. By 2018, it reached two hundred and twenty-four million. The growth trajectory validated two things: the depth of unmet need in PDP and the effectiveness of ACADIA's specialty sales model.
Then, on April 9, 2018, CNN published a bombshell investigative report that threatened to undo everything. The headline: "Reports of death spark concern about Parkinson's drug."
The report, titled "Reports of death spark concern about Parkinson's drug," made several explosive allegations. More than seven hundred deaths had been reported to the FDA's Adverse Event Reporting System since NUPLAZID's approval. The FDA's own medical reviewer had recommended against approval and was overruled. The drug was approved based on a single positive six-week trial of approximately two hundred patients, after three previous trials had failed. And the measurement scale used in the successful trial, the SAPS-PD, "had not been validated."
The stock plummeted more than twenty-two percent in a single trading session. The Institute for Safe Medication Practices published QuarterWatch reports flagging NUPLAZID as a safety signal, calling for stronger warnings. And in September 2018, ACADIA disclosed receiving a civil investigative demand from the Department of Justice under the False Claims Act, requesting documents related to NUPLAZID sales and marketing practices. CNN had also reported that roughly a quarter of the top Medicare prescribers of NUPLAZID were listed as paid ACADIA consultants.
This was the second existential crisis.
A widely watched cable news investigation accusing your only product of killing patients. A DOJ investigation into your marketing practices. Safety watchdog groups calling for stronger warnings. For a company with a single commercial product, this was an extinction-level event.
ACADIA's response was methodical. The company pointed to a critical piece of context that the CNN report had largely ignored: PDP patients are elderly, frail, and have advanced neurodegenerative disease. They have an inherently high mortality rate. ACADIA's own analysis showed that the mortality rate among NUPLAZID patients was twelve and a half deaths per hundred patient-years, less than half the twenty-eight deaths per hundred patient-years observed in the general PDP population not taking the drug. In other words, patients on NUPLAZID were dying at lower rates than patients not on it, though this comparison had obvious limitations and was not from a randomized trial.
The FDA took six months to complete its own review. On September 20, 2018, it issued a formal Drug Safety Communication that was about as close to vindication as ACADIA could have hoped for: "FDA did not identify any new or unexpected safety findings with Nuplazid, or findings that are inconsistent with the established safety profile currently described in the drug label." The benefit-risk profile remained favorable.
The FDA's review did note some "potentially concerning prescribing patterns," including concomitant use of NUPLAZID with other antipsychotics or QT-prolonging medications. But the bottom line was clear: the drug stayed on the market, with no new restrictions. Key opinion leaders like Dr. Kevin Black of Washington University published measured rebuttals of the CNN investigation, and the Parkinson's Foundation issued supportive statements after the FDA's review.
The damage was real but not fatal.
ACAD stock fell more than forty percent over the course of 2018 before recovering after the FDA's all-clear. The DOJ investigation would continue but ultimately did not result in enforcement action. The episode left lasting reputational scars and heightened scrutiny, but it also demonstrated something important about ACADIA: the company knew how to survive a crisis.
The 2018 controversy also revealed a fundamental tension in rare disease and specialty drug markets. Adverse event reporting databases like FAERS capture every death that occurs while a patient is on a medication, regardless of whether the drug caused the death. For a drug prescribed to elderly patients with advanced Parkinson's disease, a population with inherently high mortality rates, the raw numbers will always look alarming. The key question is comparative: are patients dying at higher rates on the drug than they would without it? ACADIA's data suggested the answer was no, and the FDA's independent review confirmed that the benefit-risk profile remained favorable. But the gap between statistical reality and media narrative is vast, and navigating that gap remains one of the most challenging aspects of commercializing drugs for vulnerable patient populations.
VII. Expansion Strategy & Pipeline Diversification (2017-2021)
Coming out of the 2018 safety controversy, ACADIA faced a strategic question that every single-product pharmaceutical company must eventually answer: what happens if your one product stumbles? The CNN episode had made that question existential.
The most obvious path to diversification was label expansion. PDP, while a meaningful market, represented perhaps a hundred thousand treated patients in the United States. Dementia-related psychosis, or DRP, was estimated to be ten times larger. Approximately twenty to seventy percent of all dementia patients experience hallucinations or delusions at some point, and with the aging population driving dementia prevalence ever higher, DRP represented a multi-billion-dollar opportunity.
ACADIA designed the HARMONY study, a Phase III relapse prevention trial that represented a genuinely innovative approach to proving efficacy in this population. Instead of the traditional parallel-group design that had failed in earlier trials, HARMONY used a "randomized withdrawal" methodology. All patients first received pimavanserin for twelve weeks in an open-label stabilization period. Those who responded well, about sixty-two percent of eligible patients, were then randomized to either continue pimavanserin or switch to placebo for up to twenty-six weeks. The primary endpoint was time to relapse.
The results, announced in September 2019 when an independent Data Monitoring Committee stopped the trial early for positive efficacy, were striking. Pimavanserin reduced the risk of psychosis relapse by nearly three-fold compared to placebo. Only thirteen percent of patients on pimavanserin relapsed, versus twenty-eight percent on placebo. The Parkinson's disease dementia subgroup was particularly dramatic: only one of thirteen patients on drug relapsed versus nine of seventeen on placebo, a ninety-five percent reduction in relapse risk.
ACADIA submitted its supplemental NDA in July 2020, seeking a broad DRP indication across multiple dementia subtypes including Alzheimer's, Lewy body dementia, Parkinson's disease dementia, vascular dementia, and frontotemporal dementia.
The stock surged to nearly fifty-eight dollars on July 7, 2020 — its all-time high. The market was pricing in a multi-billion-dollar DRP franchise.
Then the FDA delivered a gut punch. In April 2021, the agency issued a Complete Response Letter rejecting the broad DRP indication. The FDA acknowledged the positive overall results but stated it could not approve the application because of insufficient evidence of efficacy in some specific dementia subtypes, particularly vascular dementia and frontotemporal dementia, where too few patients had been enrolled to demonstrate statistical significance.
ACADIA regrouped and resubmitted in February 2022 with a narrowed focus on Alzheimer's disease psychosis specifically. But in June 2022, the advisory committee voted nine to three against the efficacy case. In August 2022, the FDA issued a second CRL, again declining to approve. The agency recommended that ACADIA conduct an additional clinical trial in Alzheimer's disease psychosis.
Two regulatory rejections for the DRP expansion. The stock collapsed from its all-time high of fifty-eight dollars to below twenty dollars.
The one-product-company problem was now acute.
The DRP saga illustrates one of the most frustrating dynamics in drug development: a drug can work in clinical trials, demonstrate statistically significant benefit, get published in the New England Journal of Medicine, and still get rejected by the FDA if the evidence does not meet the agency's specific requirements for subgroup consistency and evidence quality. ACADIA's HARMONY trial was a clinical success; it was a regulatory failure. The distinction matters enormously, and understanding it is key to evaluating ACADIA's next attempt at the DRP market through remlifanserin. ACADIA needed a second act, and fortunately, it had been building one.
VIII. The Trofinetide Pivot & Rett Syndrome Opportunity (2018-2023)
Rett syndrome is one of the cruelest diseases in medicine. It overwhelmingly affects girls, caused by spontaneous mutations in the MECP2 gene on the X chromosome. Babies appear entirely normal at birth, develop normally for six to eighteen months, and then begin a devastating regression. They lose the ability to speak, to use their hands purposefully, to walk normally. Stereotyped hand movements, hand wringing and clapping, replace intentional gestures. Seizures develop in roughly eighty percent of patients. Breathing becomes irregular. The patients, trapped in bodies that will not cooperate, remain cognitively aware enough to suffer.
There are approximately six to nine thousand Rett patients in the United States and fifteen to twenty thousand globally. The disease primarily affects girls because boys with MECP2 mutations rarely survive infancy.
Until 2023, there was not a single FDA-approved treatment. Not one.
In August 2018, just months after the CNN safety crisis, ACADIA made what would prove to be one of its most consequential strategic decisions. It signed an exclusive license agreement with Neuren Pharmaceuticals, an Australian company, for North American rights to trofinetide, a synthetic analogue of a naturally occurring brain protein fragment. Neuren received ten million dollars upfront and was eligible for up to four hundred and fifty-five million in milestone payments.
Trofinetide is what scientists call a first-in-class medication. It is a synthetic version of glycine-proline-glutamate, or GPE, a naturally occurring fragment of insulin-like growth factor 1 found in the brain. The drug works through multiple pathways simultaneously: it reduces the neuroinflammation caused by overactive brain immune cells, normalizes synaptic structure, and restores neuronal signaling. Think of it as quieting a storm of inflammation in the brain while simultaneously rebuilding the damaged circuitry.
The clinical development story was remarkably clean for such a difficult disease. Neuren's Phase II studies, including a study in eighty-two girls and adolescents, showed that the highest dose achieved statistically significant improvements on Rett-specific behavioral measures and clinician-assessed global improvement. ACADIA took over for the pivotal Phase III trial, called LAVENDER, which enrolled a hundred and eighty-seven females aged five to twenty with Rett syndrome.
Both co-primary endpoints met statistical significance. The Rett Syndrome Behaviour Questionnaire showed meaningful improvement over placebo, and the Clinical Global Impression-Improvement scale confirmed that investigators observed real clinical benefit. Trofinetide received Fast Track designation, Orphan Drug designation, Rare Pediatric Disease designation, and Priority Review.
On March 10, 2023, the FDA approved DAYBUE, making trofinetide the first and only FDA-approved treatment for Rett syndrome in adults and pediatric patients two years of age and older.
The approval also came with a Rare Pediatric Disease Priority Review Voucher, which ACADIA would later sell for a hundred and fifty million dollars in December 2024. For a company that had spent years on the financial edge, that voucher was a hundred-and-fifty-million-dollar bonus for doing something nobody else had managed to do.
The launch came with significant challenges. DAYBUE is dosed as an oral solution based on body weight, and the annual cost ranges from roughly three hundred and eighty-five thousand to over nine hundred thousand dollars at list price, depending on patient weight. The average net realized cost to payers was approximately three hundred and seventy-five thousand dollars per year. And the side effect profile was demanding: more than eighty percent of patients in clinical trials experienced diarrhea, with about fifteen to twenty percent discontinuing treatment because of it.
Despite these hurdles, the launch exceeded expectations.
DAYBUE generated a hundred and seventy-seven million dollars in its first year, ramped to three hundred and forty-eight million in 2024, and was on track for three hundred and eighty-five to four hundred million in 2025. By the third quarter of 2025, over a thousand unique patients were receiving DAYBUE shipments per quarter. The company was finally a two-product enterprise.
In July 2023, ACADIA expanded its Neuren partnership, acquiring ex-North American rights to trofinetide and global rights to Neuren's pipeline candidate NNZ-2591 for a hundred million dollars upfront plus royalties and milestones. The European Marketing Authorization Application was submitted, though in February 2026 the EMA's committee issued a negative trend vote, a setback that ACADIA planned to challenge through re-examination.
A December 2025 approval of DAYBUE STIX, a powder formulation available in three weight-based dosage strengths, addressed some of the practical challenges of the original liquid formulation, offering improved convenience and potentially better tolerability. The original oral solution required patients to consume large daily volumes of liquid, a practical challenge for patients with swallowing difficulties, which is common in Rett syndrome. STIX was expected to be broadly available by early second quarter 2026.
The Neuren royalty structure provides important context for understanding DAYBUE's economics. Neuren receives tiered royalties on North American net sales: ten percent on the first two hundred and fifty million, twelve percent up to five hundred million, fourteen percent up to seven hundred and fifty million, and fifteen percent above that threshold. There are also sales milestone payments, including fifty million dollars each at the two hundred and fifty and five hundred million dollar thresholds, and a hundred million at seven hundred and fifty million. In 2024, Neuren received fifty-six million Australian dollars in royalties plus an eighty-point-five million Australian dollar milestone payment when annual net sales crossed two hundred and fifty million. These royalty obligations are a meaningful cost of goods for DAYBUE, but they are the price of accessing a drug that ACADIA did not discover internally, and few investors would argue it was not worth it.
IX. Dementia-Related Psychosis: The Second Act (2021-2024)
After two CRLs for the broad DRP and narrower Alzheimer's disease psychosis indications, ACADIA's DRP ambitions appeared stalled. But the company found an important, if partial, victory through a different route.
In September 2023, the FDA updated the NUPLAZID prescribing label to clarify that pimavanserin is approved for the treatment of psychosis in Parkinson's disease patients with or without dementia. This was a label clarification, not a new indication approval, but it was commercially significant. It explicitly acknowledged that the pivotal Phase III study had included Parkinson's patients with coexisting dementia, and that the drug was appropriate for this overlapping population.
This clarification broadened the addressable market within NUPLAZID's existing indication. Many Parkinson's disease patients develop dementia as their disease progresses, and the updated label gave physicians greater confidence to prescribe NUPLAZID to these patients without feeling they were venturing off-label.
The broader DRP market remains the biggest commercial prize in ACADIA's future, and it dwarfs everything the company has done so far.
An estimated thirteen million nine hundred thousand Americans aged sixty-five and older are projected to be diagnosed with dementia by 2060, with approximately thirty percent of Alzheimer's patients experiencing psychosis. No drug is currently approved for this indication, making it one of the largest unmet needs in CNS medicine.
ACADIA is now pursuing this opportunity not through pimavanserin but through a next-generation molecule called remlifanserin, designated ACP-204. This is a novel, highly selective 5-HT2A receptor inverse agonist designed to improve upon pimavanserin's profile. The Phase II RADIANT study in Alzheimer's disease psychosis is underway, with top-line results expected between August and October 2026. The program uses a seamless Phase 2/3 design, meaning that if Phase II succeeds, the same trial infrastructure can transition directly into confirmatory Phase III studies.
The competitive landscape is also evolving. Bristol Myers Squibb's Cobenfy, a muscarinic receptor agonist currently approved for schizophrenia, is being studied in Alzheimer's disease psychosis through the ADEPT Phase III program. BMS has identified site irregularities at some trial locations, and results are expected by end of 2026. If both drugs succeed, the DRP market could become a multi-billion-dollar competitive battleground.
NUPLAZID revenue growth has remained steady despite the DRP setbacks, a testament to the underlying strength of the PDP franchise.
Revenue grew from four hundred and forty-two million in 2020 to five hundred and forty-nine million in 2023, and then accelerated to six hundred and nine million in 2024. The third quarter of 2025 marked a record quarter for NUPLAZID at a hundred and seventy-seven and a half million, representing twelve percent year-over-year growth including nine percent volume growth. Full-year 2025 NUPLAZID guidance was raised to six hundred and eighty-five to six hundred and ninety-five million.
X. The Business Model & What Makes ACADIA Different
ACADIA operates as a pure-play CNS specialist, and this focus has been both its greatest strength and its most persistent vulnerability. In an industry where the largest pharmaceutical companies maintain sprawling portfolios across oncology, immunology, cardiovascular disease, and infectious disease, ACADIA has bet everything on the brain.
The company's R&D philosophy starts with mechanism rather than indication. Instead of asking "what disease should we treat?" ACADIA asks "what biological mechanism do we understand deeply enough to exploit?" The R-SAT platform identified 5-HT2A inverse agonism as a promising mechanism in the late 1990s. It then took over a decade to find the right disease in which to apply it. This is the opposite of how most pharmaceutical companies work, where a disease is chosen first and then a drug is designed for it.
The commercial model is pure specialty. ACADIA maintains a sales force of approximately two hundred and ten representatives, expanded roughly thirty percent from earlier configurations, using AI-driven "precision execution" to optimize targeting. The sales force is bifurcated between community/office settings, where neurologists and movement disorder specialists practice, and institutional settings like nursing homes and long-term care facilities. NUPLAZID holds low-twenties market share penetration in the community setting and mid-twenties in long-term care.
The manufacturing model is entirely outsourced. Patheon Pharmaceuticals and Catalent Pharma Solutions manufacture NUPLAZID under contract, providing redundancy without the capital burden of owned facilities. This asset-light approach is typical of mid-sized specialty pharma and allows ACADIA to concentrate its capital on R&D and commercialization.
The financial profile has undergone a dramatic transformation, from decades of losses to sustained profitability. ACADIA crossed the billion-dollar revenue milestone in 2025, with full-year guidance of one point zero seven to one point zero nine five billion dollars. The company achieved its first full year of GAAP profitability in 2024, reporting net income of two hundred and twenty-six and a half million, though this included a one-time gain of roughly a hundred and forty-six million from the priority review voucher sale. Even excluding that, the underlying operations turned profitable.
The balance sheet is exceptionally clean. As of late 2025, ACADIA held approximately eight hundred and forty-seven million dollars in cash and short-term investments, with zero debt. Total shareholder equity stood at roughly nine hundred and seventeen million, and short-term assets of one point one billion comfortably exceeded the combined short and long-term liabilities of about four hundred and fourteen million. This gives the company substantial firepower for pipeline investments and potential acquisitions.
The leadership transition in September 2024 marked a meaningful shift in strategic posture, and it may prove to be the most consequential personnel decision since Uli Hacksell was hired in 2001. Catherine Owen Adams, who succeeded Steve Davis as CEO, brought a strikingly different profile to the role. Where Davis was the CPA-lawyer who built the commercial infrastructure from zero, Adams was a big-company commercial leader. Her most recent role was as Senior Vice President and General Manager of Bristol Myers Squibb's U.S. business, where she oversaw twenty billion dollars in revenue and more than three thousand employees. Before BMS, she spent twenty-five years at Johnson & Johnson, ultimately serving as President of Janssen Immunology U.S. She began her career in R&D and manufacturing at AstraZeneca in the UK and holds a pharmacy degree from the University of Manchester.
Adams brought a personal connection to ACADIA's mission: both of her parents have Alzheimer's disease. She has been blunt about her ambitions, stating publicly that she "didn't come into this job to putter along at a three-billion-dollar market cap" and signaling a more assertive approach to business development. The company also bolstered its strategic capabilities by appointing Konstantina Katcheves as Senior Vice President, Chief Business and Strategy Officer, to lead global partnerships and transactions.
Beyond NUPLAZID and DAYBUE, ACADIA's pipeline has expanded significantly under new leadership, with the company disclosing nine active programs at its inaugural R&D Day in June 2025 and anticipating seven Phase II or Phase III study initiations during 2025 and 2026. A collaboration with Stoke Therapeutics, signed in January 2022 for sixty million dollars upfront, is developing RNA-based medicines using Stoke's proprietary TANGO platform. The partnership targets SYNGAP1 syndrome and Rett syndrome through antisense oligonucleotides that restore missing protein production. A separate collaboration with Axcelead Drug Discovery Partners, a Japanese drug discovery company spun out of Takeda's platform, is supporting multiple neuroscience discovery projects. The company's Phase III COMPASS trial of intranasal carbetocin for Prader-Willi syndrome failed to meet its primary endpoint in September 2025 and was discontinued, a reminder that not every pipeline bet pays off. But the pipeline has also been enriched with entirely new molecules. ACP-211, a selectively deuterated form of R-norketamine, entered Phase II for major depressive disorder in the fourth quarter of 2025, representing a potential oral antidepressant with the efficacy of ketamine but without the sedation and dissociation. ACP-711, a selective GABAA alpha-3 modulator licensed from Saniona in November 2024, completed Phase I with a clean safety profile and is headed into Phase II for essential tremor in 2026. And ACP-271, a first-in-class GPR-88 agonist, entered first-in-human studies in early 2026. The company estimates its full pipeline potential at roughly eleven billion dollars in peak sales, though this is illustrative rather than guidance.
XI. The Competitive Landscape & Future Threats
ACADIA's competitive position is unusual for a pharmaceutical company: it currently has no direct approved competitors in either of its marketed indications. NUPLAZID remains the only FDA-approved treatment for Parkinson's disease psychosis, nine years after its approval. DAYBUE remains the only FDA-approved treatment for Rett syndrome, nearly three years after launch. This kind of monopoly position in approved therapies is rare and valuable, but it invites both opportunity and risk.
In PDP, the de facto competition comes from off-label use of atypical antipsychotics, primarily quetiapine and clozapine. These drugs are cheap, widely available, and familiar to physicians, but they carry significant motor side effects that make them problematic for Parkinson's patients. The off-label alternatives serve as a price anchor and a reminder that ACADIA must continuously demonstrate superior outcomes to justify NUPLAZID's premium pricing.
In the broader DRP market, the most serious competitive threat comes from Bristol Myers Squibb's Cobenfy, which works through a completely different mechanism, muscarinic receptor agonism. If the ADEPT trials succeed, Cobenfy could reach the DRP market around the same time as ACADIA's remlifanserin, setting up a head-to-head commercial battle in a market worth potentially several billion dollars annually.
ACADIA's patent position is stronger than many investors realize. The key formulation patent for NUPLAZID's thirty-four milligram capsule was upheld through August 2038 after ACADIA won a patent challenge from Aurobindo Pharma in U.S. District Court, a victory later affirmed by the U.S. Court of Appeals for the Federal Circuit. The composition of matter patent expires in 2030, and a method of use patent extends to 2037, but the formulation patent provides the most commercially relevant protection. Generic entry is not expected until August 2038.
For DAYBUE, the picture is even stronger. The new chemical entity exclusivity extends through March 2028, the indication exclusivity runs through March 2030, and the last expiring patent plus exclusivities push the estimated generic launch date to July 2042. Orphan drug designation provides additional market exclusivity protection.
The M&A question looms over ACADIA, and it cuts both ways.
In January 2017, Pfizer was reportedly in talks to acquire the company at forty-four dollars per share, a deal that never materialized. With the stock trading in the mid-twenties as of early 2026 and a market cap around three point seven billion, ACADIA trades at a significant discount to its 2020 peak and arguably below the value of its commercial and pipeline assets. Whether the company becomes a buyer or a seller will likely depend on the remlifanserin Phase II data expected in mid-2026.
One factor that does not get enough attention is ACADIA's relationship with patient advocacy organizations. In rare diseases, these groups are not just nice-to-have; they are essential partners in patient identification, clinical trial recruitment, and market development. The International Rett Syndrome Foundation played a critical role in identifying trofinetide's potential as early as 2012. ACADIA has established a Rett Parents Council to maintain direct communication with families and employs dedicated patient advocacy liaisons. In PDP, the Parkinson's Foundation, American Parkinson Disease Association, Davis Phinney Foundation, and Michael J. Fox Foundation have all featured NUPLAZID in their patient education materials. These relationships take years to build and represent a form of competitive advantage that does not appear on any balance sheet.
XII. Strategic Analysis: Porter's 5 Forces
Threat of New Entrants: Moderate. Developing a CNS drug from scratch takes over a decade and typically costs more than a billion dollars. The regulatory expertise required to navigate FDA advisory committees, design psychosis measurement scales, and manage post-market surveillance creates meaningful barriers to entry. But Big Pharma companies like Bristol Myers Squibb have the resources and scientific capability to enter these markets when they sense an opportunity, as the Cobenfy program demonstrates.
Bargaining Power of Suppliers: Low. ACADIA's outsourced manufacturing model relies on established contract manufacturers. The active pharmaceutical ingredient for pimavanserin is not particularly complex, and multiple CMOs could produce it. Patheon and Catalent provide current manufacturing, with adequate redundancy to prevent supply disruption.
Bargaining Power of Buyers: Moderate to High. Payers, particularly Medicare and Medicaid, have significant leverage over pricing, especially for high-cost specialty drugs. Prior authorization requirements can slow uptake and create administrative friction. However, NUPLAZID's position as the only approved treatment in its indication limits payer ability to push patients toward alternatives. The tension between monopoly pricing power and payer resistance is an ongoing negotiation.
Threat of Substitutes: Moderate. Off-label antipsychotics are cheaper but carry worse side effects, and crucially, none have FDA approval for PDP. Non-pharmacological interventions, such as reducing dopaminergic medication doses, adjusting lighting, and behavioral approaches, offer modest benefit but are insufficient for moderate-to-severe psychosis. Future gene therapies or disease-modifying treatments for Parkinson's and Alzheimer's could eventually reduce the psychosis patient population, but these are decades away from widespread availability. Digital therapeutics, an emerging category, are exploring VR-based and app-based interventions for psychiatric symptoms but remain in early stages. The most immediate substitute threat comes from payer-mandated step therapy requiring patients to fail cheaper alternatives before accessing NUPLAZID, a practice that forces patients through the very drugs that worsen their motor symptoms before they can access the one drug designed specifically for their condition.
Competitive Rivalry: Currently Low, Increasing. NUPLAZID has had the PDP market to itself for nine years. As patent expiration approaches and the DRP market opens up, rivalry will intensify. The next two years will be critical in determining whether ACADIA can maintain its dominant position or faces meaningful competition from both branded and eventually generic entrants.
XIII. Strategic Analysis: Hamilton's 7 Powers
Scale Economies: Moderate. ACADIA benefits from fixed-cost leverage in its R&D and regulatory functions. The deep expertise in 5-HT2A biology, psychosis measurement scales, and FDA interaction protocols was built over decades and can be amortized across multiple indications. The sales force generates increasing returns as indications expand because the same neurology call points can be targeted for multiple products.
Network Economies: Low. Pharmaceutical companies generally do not benefit from network effects. However, ACADIA's growing real-world evidence database from NUPLAZID prescriptions creates a data asset that could inform future clinical development and regulatory submissions. Patient registries in Rett syndrome build a similar informational advantage.
Counter-Positioning: Low. ACADIA is not disrupting an incumbent's business model in the classic counter-positioning sense. Large pharma companies could pursue the same indications without cannibalizing existing products. The reason most large companies have not been in PDP is the small market size and high development risk, not a structural inability to compete.
Switching Costs: Moderate to High. Physicians who have successfully managed patients on NUPLAZID are reluctant to switch, particularly given the vulnerable nature of PDP patients and the lack of approved alternatives. Caregivers who have found stability with NUPLAZID are even less inclined to change. In Rett syndrome, the rarity of the disease and the complexity of the dosing regimen create additional inertia. These switching costs will weaken as patents expire and generics become available, but they provide meaningful near-term protection.
Branding: Moderate to High. NUPLAZID has strong brand recognition within the PDP specialist community. The company's direct-to-consumer campaigns have built caregiver awareness. ACADIA's scientific reputation, built through published clinical trials in top journals including the New England Journal of Medicine, lends credibility. The 2018 safety controversy tested this brand but ultimately, the FDA's vindication strengthened it.
Cornered Resource: Moderate to High. This is ACADIA's strongest power. Three decades of CNS-focused research have created deep expertise in serotonin receptor pharmacology, psychosis measurement, and regulatory navigation in rare neurological diseases. The company's key opinion leader relationships, built over years of clinical collaboration, represent a network that competitors cannot easily replicate. The institutional knowledge of how to design and execute clinical trials in populations that are notoriously difficult to study, elderly PDP patients, children with Rett syndrome, is a genuine cornered resource.
Process Power: Moderate. ACADIA has developed a refined playbook for clinical development, regulatory filing, and specialty commercialization that has been validated across two drug approvals and multiple indications. The ability to learn from trial failures and redesign approaches, as demonstrated by the pivot from Study 012 to Study 020, represents accumulated process knowledge that would take competitors years to replicate.
In summary, ACADIA's competitive moat is primarily built on cornered resource — deep expertise, relationships, and regulatory knowledge — and switching costs. As patent expiration approaches in the early-to-mid 2030s for composition of matter and 2038 for formulation, these softer moats become increasingly critical.
The question is whether the company can translate three decades of CNS expertise into a sustainable multi-product franchise before the patent clock runs out on its two existing drugs. The pipeline expansion under Adams suggests management understands this imperative.
XIV. Bear vs. Bull Case & Investment Considerations
The Bull Case.
ACADIA is on the cusp of a revenue transformation. The NUPLAZID label clarification for PD patients with dementia has re-accelerated prescription growth, with the third quarter of 2025 showing record revenue and double-digit growth. DAYBUE continues to penetrate the Rett syndrome market, with over a thousand patients receiving shipments per quarter and the new STIX powder formulation potentially improving tolerability and adherence. If remlifanserin succeeds in Phase II for Alzheimer's disease psychosis, it opens a market that ACADIA estimates at roughly four billion dollars in peak sales. Combined NUPLAZID and DAYBUE peak sales are projected at one and a half to two billion dollars, with full pipeline potential reaching eleven billion. The company is debt-free with nearly eight hundred and fifty million in cash, profitable, and run by a new CEO with a mandate to grow through business development. Patents extend to 2038. And at a market cap of roughly three point seven billion against a billion in revenue, the stock trades at a modest multiple for a growing specialty pharma company with significant pipeline optionality.
The Bear Case.
ACADIA remains heavily dependent on NUPLAZID, which accounts for roughly two-thirds of revenue. Composition of matter patents expire in 2030, and while formulation patents extend to 2038, generic manufacturers will begin preparation well before expiration. The DRP expansion through remlifanserin is unproven, and Phase II failure in mid-2026 would be a significant setback, particularly if BMS's Cobenfy succeeds and claims the market. DAYBUE's eighty percent diarrhea rate and high discontinuation rates limit patient retention and long-term revenue potential. The EMA's negative trend vote for trofinetide in Europe raises questions about international expansion. The 2018 safety controversy, while resolved by the FDA, left a residual perception overhang. And ACADIA's market cap has already recovered from its 2024 lows, meaning much of the near-term optimism is priced in.
Key Metrics to Track.
For investors following ACADIA, two metrics matter above all others. First, NUPLAZID new-to-brand prescriptions, or NBRx, which measures the rate at which new patients are starting the drug. This is the leading indicator of whether NUPLAZID revenue growth can continue or is approaching a ceiling. The nine percent volume growth reported in Q3 2025 is encouraging, but any deceleration in NBRx trends would be an early warning signal. Second, DAYBUE active patient count, specifically the number of unique patients receiving shipments per quarter. This metric captures both new patient starts and, critically, retention, which is challenged by the drug's tolerability profile. The trajectory from roughly nine hundred and fifty patients in early 2025 to over a thousand in Q3 2025 needs to continue accelerating for DAYBUE to reach its peak sales potential.
XV. Lessons for Founders & Investors: The ACADIA Playbook
ACADIA's three-decade journey from a Vermont lab to a billion-dollar revenue company offers lessons that transcend the biotech sector.
Survival is the prerequisite to success.
ACADIA nearly died multiple times: during the 2008 financial crisis with the stock below a dollar, after multiple Phase III failures, after the 2018 CNN investigation. Each time, the company survived through a combination of creative financing, lean operations, and sheer tenacity. The committed equity facility from Kingsbridge, the Biovail partnership and subsequent reacquisition of rights, the methodical response to the safety controversy, these were not glamorous moves, but they kept the company alive long enough for the science to work.
Finding the right indication can take longer than developing the drug itself.
Pimavanserin was synthesized in the early 2000s. It failed in schizophrenia as a standalone treatment. It showed promise in schizophrenia as an add-on. It was studied for insomnia. It failed two Phase III trials in PDP before succeeding in a redesigned third trial. The decision to focus on Parkinson's disease psychosis, and then to rethink the trial design, measurement scale, and patient selection, was ultimately what made the difference. The molecule itself did not change; the understanding of where it belonged did.
Regulatory strategy is not a box to check; it is the strategy. The way ACADIA engaged with the FDA, from aligning on trial endpoints to obtaining Breakthrough Therapy Designation to navigating the advisory committee process, was not an afterthought. It was central to the company's success. The development of the SAPS-PD scale, controversial as it was, demonstrated deep understanding of what the FDA needed to see. The 2016 approval, the 2023 DAYBUE approval, and the 2023 NUPLAZID label clarification were all products of sophisticated regulatory thinking.
The second product is existential.
ACADIA spent seven years after NUPLAZID's approval as a one-product company. The 2018 safety crisis showed how vulnerable that position was. The trofinetide licensing deal in 2018, which cost only ten million dollars upfront, turned out to be transformative. DAYBUE's approval in 2023 diversified the revenue base and proved that ACADIA could develop and commercialize beyond PDP.
Domain expertise compounds.
The deep CNS and serotonin receptor knowledge that ACADIA built over three decades is not easily replicable. It informed the design of pimavanserin, the redesign of failed clinical trials, the development of remlifanserin, and the company's ability to evaluate and pursue the trofinetide opportunity. This accumulated expertise is the company's most durable competitive advantage. Consider the tragedy of Dr. Srdjan "Serge" Stankovic, who joined ACADIA in 2015 as head of R&D and was instrumental in the approval of both NUPLAZID and DAYBUE. He retired in 2022 and passed away in December 2023 following a prolonged illness. The scientific leadership that guided two first-in-class drug approvals is irreplaceable on a personal level, even as the institutional knowledge he helped build endures in the organization.
When to partner versus when to own.
The Biovail deal demonstrated both sides of this equation. Partnering in 2009 brought thirty million dollars and a lifeline during the financial crisis. Buying back the rights in 2010 for under nine million dollars gave ACADIA full ownership of what became a six-hundred-million-dollar-per-year product. The Neuren deal was the opposite: rather than spending years and hundreds of millions to discover trofinetide internally, ACADIA paid ten million dollars upfront and accepted a royalty structure in exchange for immediate access to a Phase III-ready asset. Both approaches worked because they matched the company's capabilities and financial position at the time.
XVI. Current State & Future Outlook (2024-2030)
ACADIA enters 2026 in the strongest position in its history. Total revenue guidance for 2025 has been raised to the one point zero seven to one point zero nine five billion dollar range, representing roughly eleven percent growth. The company is profitable, debt-free, and has nearly eight hundred and fifty million dollars in cash.
The most important catalyst on the near-term horizon is the Phase II RADIANT study readout for remlifanserin in Alzheimer's disease psychosis, expected between August and October 2026. If positive, this data could validate ACADIA's path into the DRP market through a next-generation molecule rather than the pimavanserin that the FDA twice declined to approve for that indication. ACADIA estimates remlifanserin's peak sales potential at roughly four billion dollars across Alzheimer's psychosis and Lewy body dementia psychosis.
Under new CEO Catherine Owen Adams, the company has articulated a five-year strategic vision targeting one point seven billion in combined NUPLAZID and DAYBUE sales by 2028, with the full pipeline opportunity at eleven billion. Adams has signaled a more aggressive business development posture, with the company's strong cash position enabling strategic acquisitions and licensing deals.
International expansion represents both opportunity and challenge. The EMA's negative trend vote on trofinetide in February 2026 was a setback, though ACADIA plans to request re-examination. Named patient supply programs outside the U.S. are already contributing to DAYBUE revenue, and orphan drug designations have been obtained in Japan.
The pipeline beyond NUPLAZID and DAYBUE deserves careful attention. Remlifanserin is the most important near-term catalyst, but the Stoke Therapeutics collaboration on RNA-based medicines for SYNGAP1 syndrome and Rett syndrome represents a potentially transformative longer-term opportunity. The TANGO platform uses antisense oligonucleotides to restore missing protein production in genetic diseases, and if successful, could provide disease-modifying rather than symptom-treating therapies. The Axcelead partnership is earlier stage, focused on hit-to-lead chemistry for multiple neuroscience targets, and represents ACADIA's effort to refill the discovery pipeline for the next decade.
The question investors must answer is whether ACADIA can transition from a two-product rare disease company into a sustainable, multi-billion-dollar CNS powerhouse. The remlifanserin data in 2026 will go a long way toward answering that question. Success would validate the next chapter of growth and potentially attract acquisition interest from larger pharmaceutical companies. Failure would leave ACADIA dependent on NUPLAZID and DAYBUE, both of which have meaningful but finite growth trajectories.
XVII. Epilogue: What We Learned
The ACADIA Pharmaceuticals story is, at its core, a story about the improbable persistence of scientific conviction in the face of repeated failure. From a stock trading below a dollar to a billion dollars in revenue. From a company that nearly ran out of cash to one sitting on eight hundred and fifty million. From a molecule that failed three clinical trials to the only approved drug for a devastating condition.
What surprised most in researching this story was not any single inflection point, but the sheer number of near-death experiences. Most companies that face one existential crisis do not survive. ACADIA faced at least four: the financial crisis of 2008-2009, the multiple Phase III failures, the 2018 CNN investigation, and the back-to-back FDA CRLs for DRP expansion. Each time, the company found a way forward, not through luck, but through a combination of scientific rigor, strategic pragmatism, and financial survival instinct.
The human element is easy to overlook in a story about molecules, receptors, and regulatory filings. But behind the clinical data are patients who see people who are not there, children who lost the ability to speak and use their hands, and caregivers who carry burdens that are difficult to comprehend. The fact that these patients now have FDA-approved treatment options, imperfect as they are, is not incidental to the ACADIA story. It is the point.
For founders, the lesson is about focus, expertise, and the willingness to pivot without abandoning your core thesis. For investors, it is about patience, risk tolerance, and understanding that in biotech, the regulatory catalysts are often more important than the financial ones. For everyone else, it is a reminder that the drugs we take for granted went through journeys at least as improbable as this one.
XVIII. Further Reading & Resources
Top 10 Long-Form Resources:
- ACADIA Pharmaceuticals 10-K annual reports (2015-2025), filed with the SEC, providing detailed corporate history, risk factors, and financial data.
- FDA NUPLAZID NDA review documents (2016) and the September 2018 Drug Safety Communication, available on the FDA website.
- The HARMONY study results published in the New England Journal of Medicine (July 2021), establishing the relapse prevention evidence for pimavanserin in DRP.
- The LAVENDER trial results published in Nature Medicine (2023), the pivotal data supporting DAYBUE approval for Rett syndrome.
- "On the Discovery and Development of Pimavanserin" published in the Journal of Pharmacology and Experimental Therapeutics (2014), detailing the R-SAT platform and drug design process.
- CNN's investigative report "Reports of death spark concern about Parkinson's drug" (April 2018), for understanding the safety controversy in its full context.
- Parkinson's Foundation and International Rett Syndrome Foundation patient education materials, for understanding the patient perspective.
- ACADIA's JP Morgan Healthcare Conference presentations (2016-2026), available on the investor relations website.
- The Institute for Safe Medication Practices QuarterWatch reports (2017-2019) on NUPLAZID safety signals.
- BioPharma Dive and FierceBiotech coverage of key inflection points including both FDA CRLs, the DAYBUE launch, and the CEO transition.
Books for Context:
"The Billion Dollar Molecule" by Barry Werth provides essential background on the drug development process. "Science Lessons" by Gordon Binder, the former Amgen CEO, offers relevant perspectives on building a biotech company from the ground up.
 RSS Feed
RSS Feed Spotify
Spotify Apple Podcasts
Apple Podcasts Amazon Music
Amazon Music Audible
Audible YouTube
YouTube